MDR Teil 5 - Wesentliche Neuerungen & Übergangsfristen der MDR
Mit der Veröffentlichung der Medizinprodukteverordnung (engl.: Medical Device Regulation - MDR) am 05.05.2017, kommen auf Medizinprodukte-Hersteller künftig wesentliche Neuerungen im Hinblick auf die Einhaltung regulatorischer Rahmenbedingungen und somit der Beweisführung der vom Medizinprodukt ausgehenden Wirksamkeit und Sicherheit zu.
Der nun bereits 5. Teil der MDR-Blog-Reihe beschäftigt sich neben den grundlegenden Änderungen auch mit den einzuhaltenden Übergangsfristen.
Beweggründe für die Erstellung der MDR
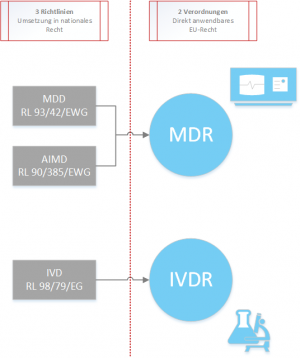
Die MDR löst die gegenwärtigen drei Richtlinien (Directives) ab:
- MDD - Medical Device Directive: RL 93/42/EWG
- AIMDD - Active Implantable Medical Device Directive: RL 90/385/EWG
- IVD - In-Vitro Diagnostica: RL 98/79/EG
MDD & AIMDD verschmelzen und werden in die MDR aufgenommen. Die In-Vitro-Diagnostika erhalten im Gegenzug eine eigenständige Verordnung (IVDR - In-Vitro Diagnostic Medical Device Regulation). Die MDR fokussiert sich stark auf internationaler Ebene entwickelte Leitlinien, um einen internationalen Angleich der Rechtsvorschriften - der weltweit zu einem hohen Niveau an Sicherheitsschutz und zum einfacheren Handel beiträgt - zu realisieren.
Gesundheitsschutzniveau für Patient & Anwender
Die Richtlinien repräsentieren die Umsetzung in nationales Recht, wohingegen die MDR einem direkt anwendbaren EU-Recht entspricht. Hauptmotivation für die Etablierung der auf EU-Ebene basierenden Verordnung ist der Wunsch nach einem reibungslos funktionierenden Binnenmarkt für Medizinprodukte. Um das hohe Gesundheitsschutzniveau für Patienten und Anwender aufrecht zu erhalten, definiert die MDR hohe Standards für Qualität und Sicherheit von Medizinprodukten und versucht gleichzeitig auch innovationsfördernd zu wirken.
Schlüsselelemente zur Verbesserung von Gesundheit und Sicherheit im Medizinprodukte-Sektor werden mit der Einführung der MDR erheblich gestärkt werden:
- Beaufsichtigung der Benannten Stellen
- Konformitätsbewertungsverfahren
- Klinische Bewertugn / Klinische Prüfung
- Vigilanz und Marktüberwachung
- Transparenz und Rückverfolgbarkeit
Verpflichtendes QMS
Für alle Hersteller von Medizinprodukten - serienmäßige Produktherstellung - fordert die MDR ein Qualitätsmanagement-System (QMS). Dieses muss alle Teile und Elemente der Organisation umfassen, die mit der Qualität der Prozesse, Verfahren und Produkte befasst sind. Diese Forderung der MDR ist unabhängig davon, ob für die Zertifizierung des Medizinproduktes der Weg der Prüfung des vollständigen QMS gewählt wurde oder nicht.
Übergangsfristen
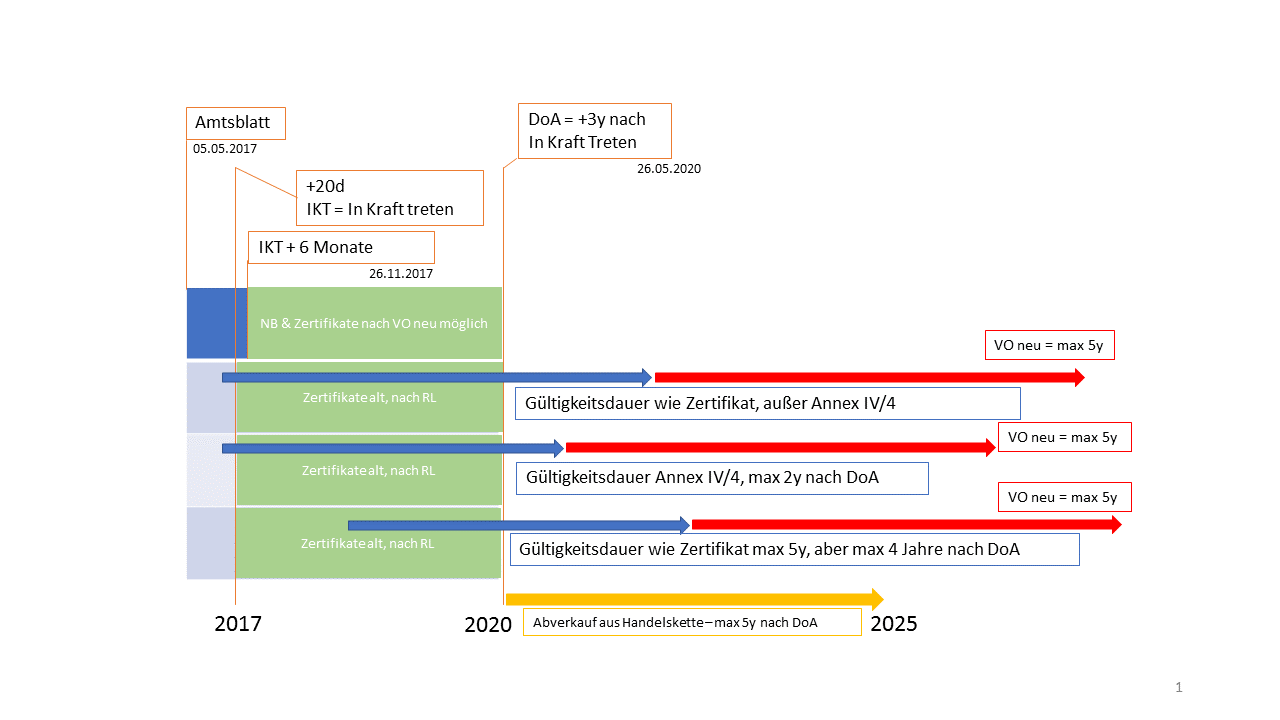 Quelle: Dr. Wolfgang Ecker
Quelle: Dr. Wolfgang Ecker
Die Veröffentlichung der MDR ist mit 05.05.2017 datiert. Am 20. Tag nach dieser Veröffentlichung tritt die MDR in Kraft (DoA - Date of Application). Ab dem 26.11.2017 (siehe Artikel 123) ist es Medizinprodukte-Herstellern möglich ihre Medizinprodukte laut Anforderungen der MDR zertifzieren zu lassen. Aufgrund der Übergangsfrist von drei Jahren, können Medizinprodukte jedoch noch bis 2020 nach den aktuell - noch - geltenden Richtlinien (Directives) zugelassen werden. Zu beachten ist in beiden Fällen - Zulassung MDR oder RL - die Gültigkeitsdauer der ausgestellten Zertifikate (siehe Grafik). Die Abverkaufsfrist - für Medizinprodukte die nach der RL legal vor oder ab dem 26.05.2020 nach Art. 120 (2) erstmals in Verkehr gebracht wurden Art 120 (4) - gilt bis exakt 27.05.2025.
Ausblick
Der nächste Blog-Artikel wird sich mit den Forderungen der MDR zum Thema “Konformitätsbewertungsverfahren” befassen.