Blog
Medizinprodukte - Oberflächen richtig reinigen
Durch Flächendesinfektionsmaßnahmen mit geeigneten Desinfektionsmitteln kann die Übertragung von Krankheiten verhindert werden. Aber nicht jede Oberfläche verträgt sich mit allen Desinfektionswirkstoffen. Neben den Händen, spielen kontaminierte Oberflächen und Gegenstände eine große Rolle bei der indirekten Übertragung von Krankheitserregern. Gerade Oberflächen mit häufigem Hand-, Haut- oder Schleimhautkontakt oder solche, die mit Körperflüssigkeiten in Kontakt kommen, bergen ein erhöhtes Infektionsrisiko, da manche Krankheitserreger sogar wochenlang auf unbelebten Flächen überleben können. Durch eine gezielte Flächendesinfektion kann dieser indirekte Infektionsweg unterbrochen und die Übertragung von Krankheiten verhindert werden. Für die Auswahl eines geeigneten Flächendesinfektionsmittels ist entscheidend, welche Arten von Mikroorganismen abgetötet werden sollen. Hierbei können Desinfektionsmittellisten wie das Expertisenverzeichnis der ÖGHMP (Österreichische Gesellschaft für Hygiene,Mikrobiologie und Präventivmedizin) oder des VAH (Verbund für Angewandte Hygiene) eine Entscheidungshilfe bieten.
Das richtige Mittel für die jeweilige Oberfläche
Aber nicht nur die mikrobielle Wirksamkeit eines Desinfektionsmittels, sondern auch dessen Materialverträglichkeit ist bei der Auswahl zu berücksichtigen. Einige, nicht gut verträglich Desinfektionswirkstoff/Material-Kombinationen sind bekannt: beispielsweise Chlorverbindungen oder Sauerstoffabspalter mit korrodierenden Metallen oder Alkohole mit Acrylglas oder Weich-PVC. In der Praxis angewendete Flächendesinfektionsmittelsind häufig Wirkstoffgemische und es zeigt sich immer wieder, dass durch den „falschen“ Einsatz von Desinfektionsmitteln vielfältige Materialschäden wie z.B. raue Stellen, Spannungsrisse, Verfärbungen, Trübung etc. entstehen können. Im schlimmsten Fall werden die aufbereiteten Gegenstände (Oberflächen, Medizinprodukte) unbrauchbar und neben hohen Reparaturkosten entstehen Diskussionen über Verantwortung, Gewährleistung oder Schadenersatz. Vor allem bei der Entwicklung von Medizinprodukten spielt die korrekte Kombination von verwendeten Materialen und Reinigungsmitteln eine wesentliche Rolle!
Ein Projekt schafft Abhilfe...
Nachdem über die Wechselwirkungen zwischen den verfügbaren Desinfektionsmitteln und den eingesetzten Materialien bzw. Oberflächen nur wenig bekannt ist, wurde von einer Expertengruppe aus den Bereichen Krankenhaus- und Industriehygiene, Analytik und Chemie, sowie Produktentwicklung und Innovation ein Projekt (HygO) zu diesem Thema bei der Österreichischen Forschungsförderungsgesellschaft eingereicht und bewilligt. Die Projektpartner (Happy Plating GmbH, HYGline GmbH, Fachhochschule OÖ – Campus Wels, mediserv Bank GmbH, Ofi Technologie & Innovation GmbH und ÖTI – Institut für Ökologie, Technik und Innovation GmbH) wollen verschiedene, gängige Oberflächen aus Metall (z. B. Aluminium, Chrom, Stahl), Kunststoff (z. B. Polypropylen, Polycarbonat, Acrylglas) und anderen Stoffen (PVC, Gummi,Glas, Keramik etc.) auf ihre Beständigkeit gegenüber ausgewählten Desinfektionsmitteln untersuchen. Neben der Beständigkeit soll auch das Alterungsverhalten dieser Oberflächen untersucht werden,um künftig auch eine langfristige Werterhaltung bei gleichzeitiger Patientensicherheit gewährleisten zu können.
... und Kompetenzen
Ein Ziel dieses von 2013 bis Oktober 2015 laufenden Projektes ist der Aufbau eines Hygienenetzwerkes, um einerseits Kompetenzen zu kombinieren und weiterzuentwickelnund andererseits Produktlösungenaufeinander abzustimmen und so materialspezifische Auswahlkriterien für Hygienekonzepte in Einrichtungen des Gesundheitswesens zu generieren. Die Ergebnisse und Erkenntnisse sollen der Industrie für Forschungs- und Entwicklungszwecke zur Verfügung gestellt werden und letztlich in die EICHYDatenbank (European Interdisciplinary Committee for Hygiene & Compatibility Testing of Medical Devices) einfließen.
Information als Entscheidungsunterstützung
EICHY stellt eine Informationsplattform dar, auf der sich Anwender kostenlos über die sichere hygienische Aufbereitung ihrer Produkte informieren können. EICHY wendet sich an medizinische Fachkräfte, Architekten, Planer und Ausstatter sowie Hersteller von (Medizin-)Produkten. Ziel von EICHY sind klare Produktdarstellungen und unmissverständliche Aufbereitungsempfehlungen, um so die Beschaffung von Medizinprodukten oder Einrichtungsgegenständen zu erleichtern und letztlich deren Funktionalität und Wert zu erhalten.
Artikel: Priv.-Doz. DI Dr. Miranda Suchomel Institut für Hygiene und Angewandte Immunologie der Medizinischen Universität Wien

Das gesamte RnB-Team freut sich, die HYGline GmbH als Kooperationspartner begrüßen zu dürfen.
Sie sind Medizinprodukte-Hersteller und möchten mehr über das Thema “Hygienische Aufbereitung von Medizinprodukten” erfahren? Dann zögern Sie nicht und kontaktieren Sie uns - office@rnb-consulting.at Gemeinsam mit unserem neuen Kooperationpartner werden wir die für Sie optimale Lösung finden!
MDR Teil 1 - Überblick über die grundlegenden Sicherheits- und Leistungsanforderungen
Es ist unbestritten, dass die kommenden europäischen Verordnungen über Medizinprodukte (MDR) bzw. In-Vitro-Diagnostika (IVDR) viele Änderungen mit sich bringen, welche die Akteure der Medizinprodukteindustrie vor große Herausforderungen stellen werden.
Pünktlich zur Veröffentlichung dieser europäischen Verordnungen möchten wir deshalb in regelmäßigen Abständen wesentliche Auszüge daraus vorstellen und die Änderungen im Vergleich zu den Vorgängerrichtlinien 93/42/EWG und 90/385/EWG diskutieren.
Die erste Blog-Reihe in diesem Kontext beschäftigt sich nun mit den Grundlegenden Sicherheits- und Leistungsanforderungen (Anhang I) der MDR. Dieser Anhang leitet sich natürlich ab von den uns bekannten Grundlegenden Anforderungen der RL 93/42/EWG bzw. der RL 90/385/EWG (Anhang I). Kurz zur Erinnerung - die Einhaltung der relevanten Anforderungen muss im Zuge des gewählten Konformitätsbewertungsverfahrens nachgewiesen werden, was im Regelfall unter Zuhilfenahme von technischen Normen geschieht. Eines nun Vorweg - diese Vorgehensweise hat sich nicht geändert. Geändert hat sich allerding der Umfang und der Detailgrad dieser Anforderungen - und das deutlich. Die grundlegenden Anforderungen der RL 93/42/EWG sind in 2 Kapitel und 13 Unterkategorien auf 9 Seiten geregelt.
- Grundlegende Anforderungen
- RL 93/42/EWG
- I. Allgemeine Anforderungen
- II. Anforderungen an die Auslegung und an die Konstruktion
- 7. chemische, physikalische und biologische Eigenschaften
- 8. Infektion und mikrobielle Kontamination
- 9. Eigenschaften im Hinblick auf die Konstruktion und die Umgebungsbedingungen
- 10. Produkte mit Messfunktion
- 11. Schutz vor Strahlungen
- 12. Anforderungen an Produkte mit externern oder interner Energiequelle
- 13. Bereitstellung von Informationen durch den Hersteller
Die Sicherheits- und Leistungsanforderungen der MDR sind in 3 Kapitel und 23 Unterkategorien auf 45 Seiten geregelt.
- Grundlegende Sicherheits- und Leistungsanforderungen
- MDR
- I. Allgemeine Anforderungen
- II. Anforderungen an die Auslegung und an die Konstruktion
- 10. Chemische, physikalische und biologische Eigenschaften
- 11. Infektion und mikrobielle Kontamination
- 12. Arzneimittel als Bestandteil, stoffliche Medizinprodukte
- 13. Produkte, zu deren Bestandteilen Materialien biologischen Ursprungs gehören
- 14. Herstellung von Produkten und Wechselwirkungen mit ihrer Umgebung
- 15. Produkte mit Diagnose- oder Messfunktion
- 16. Schutz vor Strahlung
- 17. Programmierbare Elektroniksysteme
- 18. Aktive Produkte und mit diesen verbundene Produkte
- 19. Besondere Anforderungen für aktive implantierbare Produkte
- 20. Schutz vor mechanischen und thermischen Risiken
- 21. Abgabe von Stoffen oder Energie
- 22. Schutz vor den Risiken durch Medizinprodukte, für die der Hersteller die Anwendung durch Laien vorsieht
- III. Kapitel Anforderungen an die mit dem Produkt gelieferten Informationen
- 23. Kennzeichnung und Gebrauchsanweisung
Das erste Kapitel bezieht sich jeweils auf allgemeine Anforderungen u.a. an das Risikomanagement, an die Gebrauchstauglichkeit oder an die Zuverlässigkeit entlang des Lebenszyklus. Wenig überraschend Anforderungen, welche praktisch alle Medizinprodukte erfüllen müssen.
Das zweite Kapitel liefert die Anforderungen zu produktspezifischen Eigenschaften. Erwähnenswert sind hierbei beispielsweise neue Kategorien zu
- stofflichen Medizinprodukten (12)
- Produkte, zu deren Bestandteilen Materialien biologischen Ursprungs gehören (13)
- Programmierbare Elektroniksysteme – Produkte, zu deren Bestandteilen programmierbare Elektroniksysteme gehören, und Produkte in Form einer Software (17)
- Schutz vor den Risiken durch Medizinprodukte, für die der Hersteller die Anwendung durch Laien vorsieht (22)
Einzelne Anforderungen daraus sind zwar bereits auch in der bestehenden RL 93/42/EWG abgebildet - nicht allerdings in diesem Detailgrad. Ebenso stellt das Kapitel III der MDR detailliert die Anforderungen an die Gebrauchsanweisung dar und unterscheidet sich somit maßgeblich vom Kapitel der grundlegenden Anforderung 13 der RL 93/42/EWG.
Es ist offensichtlich, dass durch die Grundlegenden Sicherheits- und Leistungsanforderungen der MDR auf aktuelle Entwicklungen reagiert wird. So werden beispielsweise Themen wie
- Stoffliche Medizinprodukte (z.B. Tränenersatzmittel)
- Software-Systeme und Spezialgebiete aus der IT (IT- Security, Wechselwirkungen mit anderen MP, …)
- Anwendungen im Home-Care Bereich
deutlich detaillierter geregelt.
Es bleibt nun abzuwarten, wie das Zusammenspiel der Normen und den Grundlegenden Sicherheits- und Leistungsanforderungen realisiert wird, da sich die harmonisierten Normen auf die vergangenen Formulierungen der RL 93/42/EWG beziehen.
Nachdem wir uns nun einen Überblick verschafft haben, werden wir uns im nächsten Blog-Artikel näher mit dem Unterkapitel I der allgemeinen Anforderungen aus der MDR beschäftigen. Im Anschluss werden wir die Klassifizierungsregeln der MDR beleuchten bzw. uns auf vergleichbare Art und Weise der IVDR annähern.
Fallzahlabschätzung bei klinischen Prüfungen - Warum ist diese so wichtig?
 Klinische Studien bzw. Prüfungen ermöglichen es, die Wirksamkeit von Interventionen auf den Menschen zu bestimmen, diagnostische Verfahren zu Beurteilen, die Prävalenz von Nebenwirkungen abzuschätzen und wissenschaftliche Hypothesen zu überprüfen. Diese sind für die Fachbereiche Medizin, Pharmakologie und Medizintechnik unerlässlich, da für die Zulassung neuartiger Therapieformen bzw. Medizinprodukte eine auf klinischen Studien basierende Datenlage gesetzlich gefordert ist.
Ein enorm wichtiger Bestandteil in der Planung klinischer Studien ist die Fallzahlabschätzung. Auch wenn es üblich ist, Parameter wie die Hauptzielgröße (Was wird untersucht?), die Art der Datenerfassung (Wie wird untersucht?), die Studiendauer (Wie lange wird untersucht?), die Studienform (In welcher Form wird untersucht?) sowie die Randomisierung (Wie erfolgt die Zuteilung der Untersuchung?) den gewünschten Anforderungen anzupassen, sollte eine Fallzahlabschätzung stets konkreten Regeln folgen.
Klinische Studien bzw. Prüfungen ermöglichen es, die Wirksamkeit von Interventionen auf den Menschen zu bestimmen, diagnostische Verfahren zu Beurteilen, die Prävalenz von Nebenwirkungen abzuschätzen und wissenschaftliche Hypothesen zu überprüfen. Diese sind für die Fachbereiche Medizin, Pharmakologie und Medizintechnik unerlässlich, da für die Zulassung neuartiger Therapieformen bzw. Medizinprodukte eine auf klinischen Studien basierende Datenlage gesetzlich gefordert ist.
Ein enorm wichtiger Bestandteil in der Planung klinischer Studien ist die Fallzahlabschätzung. Auch wenn es üblich ist, Parameter wie die Hauptzielgröße (Was wird untersucht?), die Art der Datenerfassung (Wie wird untersucht?), die Studiendauer (Wie lange wird untersucht?), die Studienform (In welcher Form wird untersucht?) sowie die Randomisierung (Wie erfolgt die Zuteilung der Untersuchung?) den gewünschten Anforderungen anzupassen, sollte eine Fallzahlabschätzung stets konkreten Regeln folgen.
Regeln der Fallzahlabschätzung
Entgegen so mancher gängigen Meinung basiert die Fallzahlabschätzung nicht auf dem Prinzip der Willkür. Typische Aussagen wie “Wir messen einmal los und wenn wir nichts finden, messen wir weiter” oder “Nehmen wir mal 10 Probanden, das wird schon genügen” haben in einer seriösen Fallzahlabschätzung nichts zu suchen. Professionelle Fallzahlabschätzungen basieren stets auf konkreten Vorerfahrungen. Dabei bilden Interventionseffekte, Grenzwerte sowie Streuungsmaße vorangegangener Untersuchungen die Berechnungsgrundlange der Fallzahlabschätzung. Sollte es keinerlei Erfahrungswerte zu neuen Intervention bzw. Produkten geben, empfiehlt es sich aus ökonomischen und ethischen Gesichtspunkten sogenannte Pilotstudien durchzuführen.
Pilotstudien
Pilotstudien sind Untersuchungen, die im Vorfeld größerer Studien durchgeführt werden. Diese kennzeichnen sich durch eine reduzierte Anzahl an teilnehmenden Personen und/oder reduzierte zeitliche Dauer. Da aus Pilotstudien konkrete Schlüsse für die Hauptstudien gezogen werden, weisen diese meist äquivalente Hauptzielgrößen, Datenerfassungsmethoden und Einschlusskriterien auf. Dies ermöglicht die Überprüfung der Akzeptanz bzw. Eignung der Messmethoden sowie die Erkennung potentieller Risiken im Vorfeld. Gegebenenfalls lässt sich dadurch auch das Studiendesign noch vor Beginn der Hauptstudie adaptieren. Gleichzeitig erlauben Pilotstudien potentielle Änderungen der Hauptzielgrößen zu ermitteln, um so eine geeignete Fallzahlabschätzung zu gewährleisten.
Bedeutung der Fallzahlabschätzung
Generell gibt es viele Gründe für die Durchführung von Fallzahlabschätzungen. Das Wahren ethischer Grundsätze sowie die zeitliche und ökonomische Optimierung klinischer Studien zählen hierbei sicher zu den wichtigsten Gründen. Im Vergleich zu rein technischen Untersuchungen, in denen z.B. der Einfluss eines Produktes auf ein anderes untersucht wird, stehen bei klinischen Studien die Einflüsse auf den Menschen im Vordergrund. Da solche Untersuchungen meist mit Einschränkungen im täglichen Leben und gewissen Restrisiken (wenn auch sehr oft sehr geringen) verbunden sind, gilt es aus ethischer Sicht immer mit einer geeigneten Anzahl an Testpersonen zu arbeiten. Werden klinische Studien beispielsweise mit zu großen Fallzahlen durchgeführt, sind mehr Menschen als notwendig mit Einschränkungen und Risiken konfrontiert. Die höheren Fallzahlen würden dabei auch zu höheren Kosten und zeitlichen Verzögerungen führen. Werden klinische Studien jedoch mit zu geringen Fallzahlen durchgeführt, wäre es nicht möglich, die Interventionseffekte wissenschaftlich zu beweisen. Da folglich weitere Studien durchgeführt werden müssten, würden erneut eine unverhältnismäßig große Anzahl an Testpersonen den negativen Auswirkungen der Studie ausgesetzt und höhere Kosten sowie zeitliche Verzögerungen verursacht werden. Da beide Situationen (zu wenig/zu viele Teilnehmer) weder ökonomisch sinnvoll sind, noch im Einklang zu ethischen Grundsätzen (Deklaration von Hellsinki) und der guten wissenschaftlichen Praxis stehen, ist es nachvollziehbar, dass die professionelle Abschätzung der Fallzahlen unabdinglich ist. Unter Berücksichtigung des zu erwartenden Effekts sowie möglicher Drop-Out- und Ausfallsraten ist es möglich, die genaue Anzahl der benötigten Probandinnen und Probanden abzuschätzen, ethische Grundsätze einzuhalten und sowohl Geld als auch Zeit zu sparen.
Dieser Beitrag wurde von unserem Kooperationspartner Bernhard Schwartz erstellt. Sie suchen zuverlässige PartnerInnen für die Abwicklung klinischer Prüfungen? Gerne steht Ihnen das R'n'B Team zur Seite und unterstützt Sie bei der Erstellung von Studienprotokollen, Ethikanträgen etc.
IEC 82304-1 / Produktsicherheit von Software-Anwendungen im Gesundheitswesen
Das stetige Wachstum von Software-Anwendungen im Gesundheitswesen, forderte die Erstellung einer explizit auf Gesundheitssoftware angepasste Norm geradezu heraus. Die IEC 82304-1 beschränkt sich nicht nur auf Medizinprodukte-Software, sondern adressiert jegliche Software-Produkte, die dazu bestimmt sind, die Gesundheit von Personen – oder auch deren Behandlung – aufrechtzuerhalten oder zu verbessern.
Wen adressiert die Norm?
Die Norm richtet sich an Hersteller reiner Software-Produkte (engl. Stand alone Software), die in der Gesundheitsbranche zum Einsatz kommen (bspw. Fitness-Apps). Demzufolge beschränkt sie sich nicht auf reine Medizinprodukte – und das ist neu! Mit dem Themengebiet der IEC 82304-1 eng in Verbindung stehend ist die Norm IEC 62304 – Medizingeräte-Software – Software-Lebenszyklus-Prozesse. Diese Norm fordert die Zusammenstellung von Prozessen, Aufgaben und Aktivitäten, die im Rahmen des Lebenszyklus-Prozesses von Medizinprodukte-Software abgewickelt werden müssen. Hauptfokus der IEC 62304 sind eingebettete Software-Komponenten (engl. Embedded Software). Da Embedded Software an sich nicht validierbar ist, sondern nur das gesamte Medizinprodukt, trifft die Norm keinerlei Aussage zu Validierungstätigkeiten. Demzufolge sind Embedded Software, medizinisch elektrische Geräte/Systeme, In-vitro Diagnostika und implantierbare Geräte von den Anforderungen der IEC 82304-1 ausgeschlossen. Für alle anderen aktiven Softwarekomponenten, die im Gesundheitswesen zum Einsatz kommen, fordert die Norm 82304-1 explizit die Durchführung von Validierungen.

Was fordert die IEC 82304-1?
Generelle Anforderungen & initiale Risikobewertung - Herstellerseitig muss klar definiert sein, welchen Zweck die Gesundheitssoftware für welche Benutzergruppe zu erfüllen hat. Das mit Sicherheitsaspekten in Verbindung stehende charakteristische Verhalten des Softwareproduktes muss analysiert und dementsprechende Risikobewertungen durchgeführt werden. Vor allem Gesundheitssoftware, die Schnittstellen zu anderen Produkten unterstützten, müssen klare Definitionen über sicherheitstechnische Realisierungen erhalten.
Nutzungsanforderung Gesundheitssoftware - Wie die Nutzungsanforderungen der Gesundheitssoftware mit deren bestimmungsgemäßen Gebrauch in Verbindung stehen, muss definiert und dokumentiert sein. Bei der Betrachtung der Nutzungsanforderungen wird die Verbindung zum Usability Engineering hergestellt. Hierbei wird die Einhaltung der IEC 62366-1:2015 empfohlen. Datenschutz- und Sicherheitsanforderungen bzgl. Authentifizierungsmethoden oder auch Integrität von Gesundheitsdaten gehen mit der Betrachtung aktuell geltender regulatorischer Bedingungen einher (Datenschutzbestimmungen) und müssen herstellerseitig klar definiert und dokumentiert werden. Die Dokumentation der Nutzungsanforderungen einer Gesundheitssoftware muss laufend aktualisiert werden. Ergebnisse aus der Verifizierung der Nutzungsanforderungen müssen aktiv in die bestehende Dokumentation einfließen. Die Verifizierung der Nutzungsanforderungen, dient der Beweisführung, dass die initial analysierten Nutzungsanforderungen herstellerseitig auch tatsächlich eingehalten und als Input für die Systemanforderungen herangezogen werden können.
Systemanforderungen Gesundheitssoftware - Die Spezifikation der Systemanforderungen dient nicht nur der Dokumentation, dass das Software-Produkt die geforderte Funktionalität, sondern in weiterer Folge auch für den Nachweis, dass der initial definierte bestimmungsgemäße Gebrauch erreicht wurde. Folgende Punkte müssen bei der Analyse der Systemanforderungen betrachtet werden: - Interoperabilität - Sprachunterstützung - Risikokontrollmaßnahmen, die auf Systemebene implementiert werden müssen’ - Anforderungen an Hard- & Software auf der die Gesundheitssoftware laufen soll
Bei der Verifizierung der Systemanforderungen ist darauf zu achten, dass sich einzelne Anforderungen untereinander nicht widersprechen und keine Mehrdeutigkeiten aufweisen. Systemanforderungen sind des Weiteren so zu definieren, dass sie eindeutig identifizierbar und einzelne Testfälle daraus generiert werden können. Zusätzlich muss eine Kompatibilität mit den zuvor definierten Nutzungsanforderungen bestehen. Für das Design des Lebenszyklus-Prozesses der Gesundheitssoftware, sind die analysierten und verifizierten Systemanforderungen als primärer Input heranzuziehen. Mit dem Design des Lebenszyklus-Prozesses eng in Verbindung stehend ist die Umsetzung der IEC 62304. Folgende Kapitel der Norm IEC 62304 i.d.g.F. sind zwingend auf Gesundheitssoftware anzuwenden: 4.2 & 4.3 + 5 – 9
Validierung der Gesundheitssoftware - Ziel der Validierung ist der geprüfte Nachweis, dass die vorab definierten Nutzungsanforderungen erfüllt sind. In erster Linie muss – auf Basis der Nutzungsanforderungen – ein Validierungsplan erstellt werden. - Definition des Validierungs-Scopes / Validierungstätigkeiten - Identifikation anwendbarer Validierungsmethoden, Input-Informationen und zugehöriger Akzeptanzkriterien - Identifikation der für die Validierung benötigten Hard- und Softwareplattformen - Spezifikation der benötigten Qualifikationen des Validierungsteams - Definition des Levels der Unabhängigkeit zwischen Validierungs- und Entwicklungsteam
Sobald der Validierungsplan freigegeben ist, kann – in der zweckmäßig vorgesehenen Umgebung - mit den Validierungsaktivitäten begonnen werden. Sofern Anomalien aufgefunden werden, müssen diese - wie in Kapitel 9 der IEC 62304 gefordert - anhand eines Problemlösungsprozesses behandelt werden. Der Abschluss aller Validierungsaktivitäten ist der Validierungsbericht. Dieser soll folgende Erkenntnisse bringen: - Validierungsergebnisse sind auf initiale Nutzungsanforderungen rückverfolgbar - Die Gesundheitssoftware erfüllt die Nutzungsanforderungen - Das Restrisiko der Gesundheitssoftware befindet sich im akzeptablen Bereich
Begleitdokumentation - Der Hersteller der Gesundheitssoftware muss Begleitpapiere zur Verfügung stellen, damit eine bestimmungsgemäße Implementierung und Anwendung erreicht werden kann. Neben der eigentlichen Gebrauchsanweisung ist auch eine technische Beschreibung des Produkts zu übermitteln. Gebrauchsanweisung: Zweckbestimmung, sichere Installation/Anwendung, sicherheitstechnische Hinweis Technische Beschreibung: Systemanforderungen an Hard- und Softwareplattform, geforderte Netzwerkkonfigurationen und Hinweis auf ggf. auftretende Risiken Sofern die Gesundheitssoftware dazu bestimmt ist in einem medizinischen IT-Netzwerk betrieben zu werden, muss der Hersteller auf sicherheitstechnische Aspekte der Netzwerkanbindung, des Netzwerkbetriebes und der Außerbetriebnahme hinweisen.
Marktbeobachtung - Mit dem Verkauf der Gesundheitssoftware endet die Verantwortung des Herstellers nicht. Laufende Aktualisierungen der Begleitdokumente müssen anlassbezogen getätigt werden. Änderungen der Software wie beispielsweise Fehlerbehebungen die Sicherheit betreffend müssen unverzüglich an den Anwender/Betreiber mitgeteilt werden.
Wie können wir Ihnen bei der Umsetzung helfen?
Damit Sie sich über die Anforderungen der IEC 82304-1 einen groben Überblick über verschaffen können, haben wir sogenannte Leitfäden erstellt – diese können Sie direkt via Mail bei uns bestellen: www.rnb-consulting.at
Neben der generellen Einführung in die Anforderungen der IEC 82304-1, stehen wir Ihnen natürlich gerne zur Verfügung, um bei der tatsächlichen Umsetzung zu helfen. GAP-Analysen bereits bestehender technischer Dokumentationen oder das gemeinsame Erstellen geforderter Spezifikationen, lassen Sie mit Sicherheit schneller ans Ziel kommen.

IEC 82304-1 & IEC 80001-1 - Könnte Kombination beider der Schlüssel für sicherere Krankenhaus IT sein?
Die kürzlich veröffentlichte Norm IEC 82304-1 stellt Anforderungen an “Health Software” (=Gesundheitssoftware), um Anwender- und Patientensicherheit, sowie Daten- und Systemsicherheit gewährleisten zu können. Mit diesen Sicherheitsanforderungen eng in Verbindung stehend sind die uns bekannte Norm EN 62304 und die Norm IEC 80001-1, die das Risikomanagement für medizinische IT-Netzwerke fordert.
IEC 82304-1 "Health Software" Produktsicherheit
Die IEC 82304-1 adressiert - mit nur wenigen Ausnahmen - jegliche Software, die im Gesundheitswesen zum Einsatz kommt und fordert den dokumentierten Nachweis der Produktsicherheit. Ausgeschlossen von diesen normativen Anforderungen sind Embedded Software, Medizinische elektrische Geräte / Systeme, In-Vitro Diagnostika und implantierbare Geräte. Stand alone Software, Medical & Health Apps sind beispielsweise sehr wohl Gegenstand dieser Norm und müssen auf potentielle Gefährdungssituationen bzw. ggf. resultierende Risiken analysiert werden. Eine Kernanforderung der IEC 82304-1 ist die nachvollziehbare Definition von Nutzungs- und Systemanforderungen an die Software, um in weiterer Folge eine repräsentative und rückverfolgbare Validierung durchführen zu können. Besonders wichtig ist die klare Definition bzw. Abgrenzung der Hard- & Software-Plattformen auf denen die Gesundheitssoftware laufen muss, um deren bestimmungsgemäßen Gebrauch gewährleisten zu können. Sofern die Gesundheitssoftware netzwerkfähig ist, muss eine kontinuierliche Hersteller-Betreiber-Kommunikation bestehen. Nur so kann eine sicherheitstechnische Netzwerkanbindung, Anwendung und Außerbetriebnahme des Produktes gewährleisten werden.
IEC 80001-1 - Risikomanagement von IT-Netzwerken, die Medizinprodukte beinhalten
Die IEC 80001-1 fordert das Risikomanagement des medizinischen IT-Netzwerkes, um zum einen drei sogenannten Schutzziele zu erreichen und zum anderen während des gesamten Lebenszyklus des IT-Netzwerkes aufrecht zu erhalten.
Die drei Schutzziele der IEC 80001-1:
- Daten- & Systemsicherheit - Schutz gegen Veränderungen der Vertraulichkeit, Verfügbarkeit und Integrität der Daten
- Sicherheit - Patienten-, Anwender- und Umweltschutz vor unvertretbaren Risiken
- Effektivität - Wirksame Patientenversorgung und optimaler interner Arbeitsablauf
Die nachstehende Grafik visualisiert den Zusammenhang der drei Schutzziele:
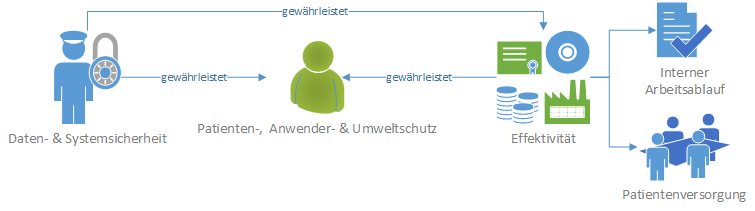
Die IEC 80001-1 ist eine reine Prozessnorm und fordert initial organisatorische Maßnahmen, wie das Aufrechterhalten der internen Kommunikationswege zwischen IT- und Medizintechnik-Abteilung, die Optimierung des Beschaffungsprozesses oder auch die Erarbeitung neuer Arbeitsanweisungen für das Klinikpersonal (z.B. Authentifizierung am Arbeitsplatz).
Krankenhaus IT ist aus vielerlei Richtungen angreifbar
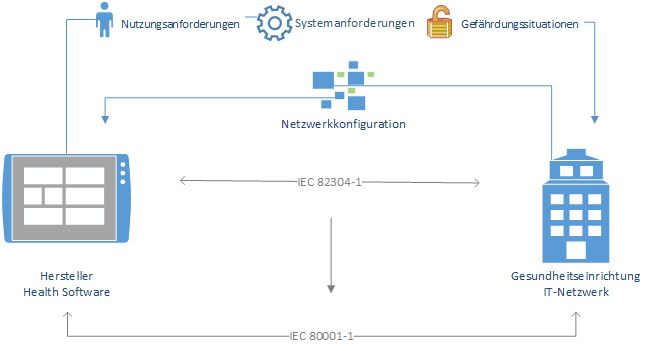
Digitalisierung in der Medizintechnik ist Innovation und Risiko zugleich. Ohne Vernetzung läuft in der Gesundheitsbranche heutzutage nur mehr wenig. Sowohl Krankenhaus-interne Arbeitsprozesse, als auch nach außen getragene Prozesse (bspw.: Ausschreibungs- und Beschaffungsprozesse) sind ohne digitale Hilfestellungen nicht mehr zu bewältigen. Umso wichtiger ist die Kommunikation zwischen Krankenhaus-Betreiber und Software-Hersteller. Der Software-Hersteller muss in seiner technischen Beschreibung Eckdaten zum bestimmungsgemäßen Gebrauch seiner Software mitliefern. Darin müssen geforderte Netzwerkkonfigurationen, vorhergesehener Informationsfluss mit anderen sich im Netzwerk befindlichen Software-Komponenten bzw. Software-Systemen und eine Liste an Gefährdungssituationen - resultierend aus der Einbettung der Gesundheitssoftware in das medizinische IT-Netzwerk - enthalten sein. In der technischen Beschreibung hat der Hersteller der Gesundheitssoftware die verantwortliche Organisation darauf hinzuweisen, dass die Ausführung der Gesundheitssoftware im medizinischen Netzwerk zu Risiken für Patient und/oder Anwender führen kann. Ggf. resultierende Risiken der Einbindung und der Anwendung der Gesundheitssoftware sind betreiberseitig zu analysieren.
Ineinandergreifen beider Normen Das Zusammenspiel beider Normen – IEC 82304-1 & IEC 80001-1 – ist für eine effektive und vor allem sichere Digitalisierung der Gesundheitsbranche notwendig. Während der Hersteller – sei es von Gesundheitssoftware oder Medizinprodukten – dem Betreiber gegenüber klar anführen muss, welche Nutzungs-, System- und Sicherheitsanforderungen für sein Produkt gelten, müssen betreiberseitig die aktuelle Netzwerkkonfiguration und dementsprechende Nutzungs- & Sicherheitsaspekte klar offengelegt werden.
Die nachstehende Grafik visualisiert einige Fakten zu den Normen IEC 82304-1 & IEC 80001-1:
- IEC 82304-1 fordert Hersteller auf, dem Anwender/Betreiber anhand von Begleitdokumenten Nutzungs-, Systemanforderungen und Gefährdungssituationen der Gesundheitssoftware zur Verfügung zu stellen
- Sofern die Gesundheitssoftware netzwerkfähig ist, wird der Betreiber des medizinischen IT-Netzwerkes aufgefordert, die Produktanforderungen mit den gegenwärtigen Netzwerkkonfigurationen abzugleichen
- IEC 80001-1 fordert eine laufende Kommunikation zwischen Hersteller und Betreiber
- Abgleich der Netzwerkkonfiguration mit Produktanforderungen
- Klare Festlegung der Verantwortlichkeiten bzgl. Wartungsarbeiten
- Herstellerseitige Meldung im Fehlerfall des Produktes
- Betreiberseitige Meldung im Falle der Änderungen/Aktualisierungen der Netzwerkbeschaffenheit –> Vergewisserung des sicherheitstechnischen Betriebes des Produktes bei Änderung der Netzwerkkonfiguration
Umsetzungsprojekte
Sie sind Hersteller einer sogenannten “Health Software“ und wollen nähere Informationen zur IEC 82304-1 oder sind Sie Betreiber einer Gesundheitseinrichtung und haben Fragen zur Umsetzungsmöglichkeiten der IEC 80001-1, dann melden Sie sich bitte direkt bei uns. Wir unterstützen Sie gerne bei der Umsetzung der normativen Rahmenbedingungen.