Blog
MDR Teil 5 - Wesentliche Neuerungen & Übergangsfristen der MDR
Mit der Veröffentlichung der Medizinprodukteverordnung (engl.: Medical Device Regulation - MDR) am 05.05.2017, kommen auf Medizinprodukte-Hersteller künftig wesentliche Neuerungen im Hinblick auf die Einhaltung regulatorischer Rahmenbedingungen und somit der Beweisführung der vom Medizinprodukt ausgehenden Wirksamkeit und Sicherheit zu.
Der nun bereits 5. Teil der MDR-Blog-Reihe beschäftigt sich neben den grundlegenden Änderungen auch mit den einzuhaltenden Übergangsfristen.
Beweggründe für die Erstellung der MDR
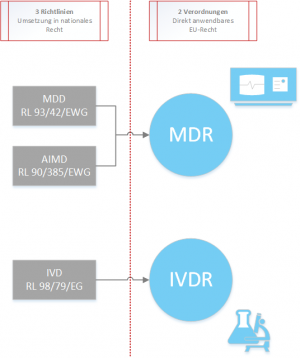
Die MDR löst die gegenwärtigen drei Richtlinien (Directives) ab:
- MDD - Medical Device Directive: RL 93/42/EWG
- AIMDD - Active Implantable Medical Device Directive: RL 90/385/EWG
- IVD - In-Vitro Diagnostica: RL 98/79/EG
MDD & AIMDD verschmelzen und werden in die MDR aufgenommen. Die In-Vitro-Diagnostika erhalten im Gegenzug eine eigenständige Verordnung (IVDR - In-Vitro Diagnostic Medical Device Regulation). Die MDR fokussiert sich stark auf internationaler Ebene entwickelte Leitlinien, um einen internationalen Angleich der Rechtsvorschriften - der weltweit zu einem hohen Niveau an Sicherheitsschutz und zum einfacheren Handel beiträgt - zu realisieren.
Gesundheitsschutzniveau für Patient & Anwender
Die Richtlinien repräsentieren die Umsetzung in nationales Recht, wohingegen die MDR einem direkt anwendbaren EU-Recht entspricht. Hauptmotivation für die Etablierung der auf EU-Ebene basierenden Verordnung ist der Wunsch nach einem reibungslos funktionierenden Binnenmarkt für Medizinprodukte. Um das hohe Gesundheitsschutzniveau für Patienten und Anwender aufrecht zu erhalten, definiert die MDR hohe Standards für Qualität und Sicherheit von Medizinprodukten und versucht gleichzeitig auch innovationsfördernd zu wirken.
Schlüsselelemente zur Verbesserung von Gesundheit und Sicherheit im Medizinprodukte-Sektor werden mit der Einführung der MDR erheblich gestärkt werden:
- Beaufsichtigung der Benannten Stellen
- Konformitätsbewertungsverfahren
- Klinische Bewertugn / Klinische Prüfung
- Vigilanz und Marktüberwachung
- Transparenz und Rückverfolgbarkeit
Verpflichtendes QMS
Für alle Hersteller von Medizinprodukten - serienmäßige Produktherstellung - fordert die MDR ein Qualitätsmanagement-System (QMS). Dieses muss alle Teile und Elemente der Organisation umfassen, die mit der Qualität der Prozesse, Verfahren und Produkte befasst sind. Diese Forderung der MDR ist unabhängig davon, ob für die Zertifizierung des Medizinproduktes der Weg der Prüfung des vollständigen QMS gewählt wurde oder nicht.
Übergangsfristen
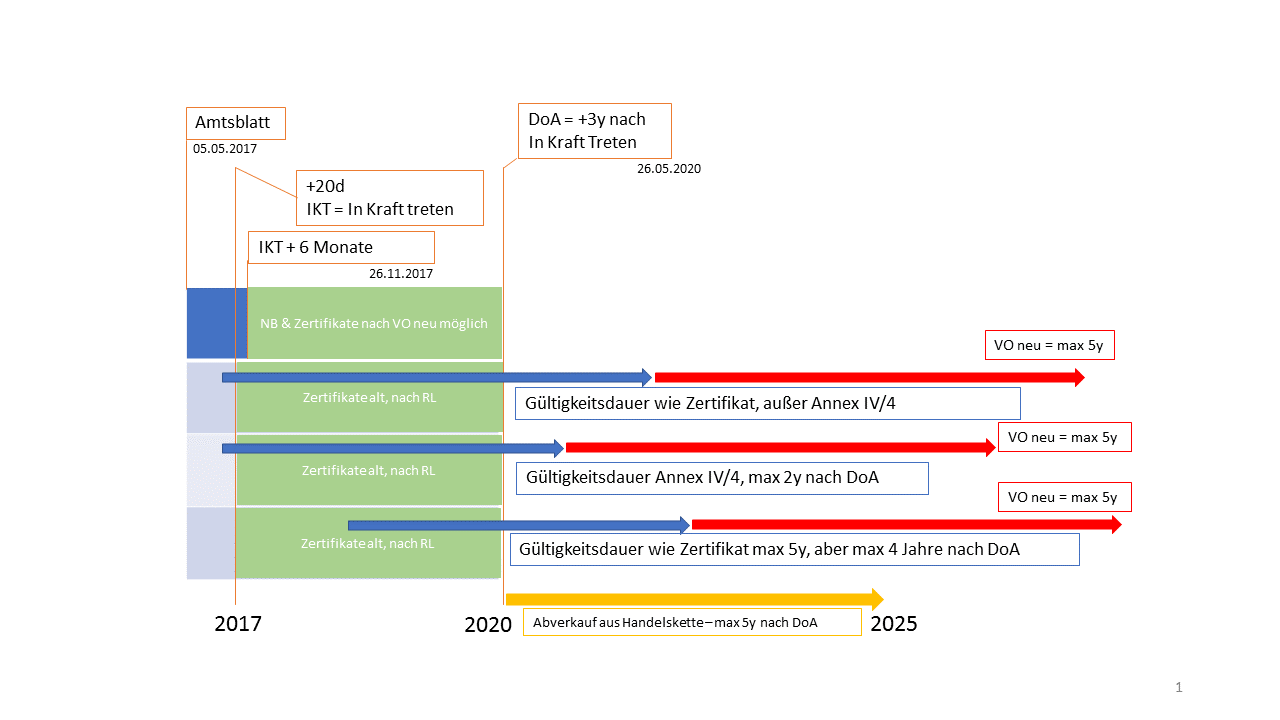 Quelle: Dr. Wolfgang Ecker
Quelle: Dr. Wolfgang Ecker
Die Veröffentlichung der MDR ist mit 05.05.2017 datiert. Am 20. Tag nach dieser Veröffentlichung tritt die MDR in Kraft (DoA - Date of Application). Ab dem 26.11.2017 (siehe Artikel 123) ist es Medizinprodukte-Herstellern möglich ihre Medizinprodukte laut Anforderungen der MDR zertifzieren zu lassen. Aufgrund der Übergangsfrist von drei Jahren, können Medizinprodukte jedoch noch bis 2020 nach den aktuell - noch - geltenden Richtlinien (Directives) zugelassen werden. Zu beachten ist in beiden Fällen - Zulassung MDR oder RL - die Gültigkeitsdauer der ausgestellten Zertifikate (siehe Grafik). Die Abverkaufsfrist - für Medizinprodukte die nach der RL legal vor oder ab dem 26.05.2020 nach Art. 120 (2) erstmals in Verkehr gebracht wurden Art 120 (4) - gilt bis exakt 27.05.2025.
Ausblick
Der nächste Blog-Artikel wird sich mit den Forderungen der MDR zum Thema “Konformitätsbewertungsverfahren” befassen.
MDR Teil 4 - Klassifizierungsregeln MDR & RL 93/42/EWG im Vergleich Teil 2
Der letzte Blog-Artikel setzte sich mit den Rahmenbedingungen für die Anwendung der Klassifizierungsregeln auseinander und stellte dabei MDR & RL 93/42/EWG gegenüber. Der nun vierte Teil der MDR-Blog-Artikel-Reihe diskutiert die Klassifizierungsregeln an sich. In der Medizinprodukte-Verordnung sind die Klassifizierungsregeln in Anhang VIII zu finden - RL 93/42/EWG definiert die Klassifizierung von Medizinprodukten in Anhang IX.
Vorweg: Der risikobasierte Ansatz der Klassifizierung eines Medizinproduktes bleibt weiterhin bestehen (Risikoklassen I - III) Grundsätzliche Neuerungen & Aufbau der Klassifizierungsregeln
Auffällig ist in erster Linie, dass sich die Anzahl der Klassifizierungsregeln von 18 (RL 93/42/EWG) auf 22 (MDR) erhöht hat. Die Aufteilung der Klassifizierungsregeln in wesentliche Anwendungscharakteristika des Medizinproduktes ist dabei jedoch gleichgeblieben – Nicht invasive Produkte, Invasive Produkte, Aktive Produkte, Besondere Regeln.

Nicht invasive Produkte: Die Klassifizierungsregeln der Produkt-Kategorie “Nicht invasive Produkte” sind - im Vergleich zur RL 93/42/EWG - in der MDR ausführlicher definiert.
- Regel 1 - gleichbleibend
- Regel 2 - Die MDR nimmt in dieser Klassifizierungsregel nun auch die Durchleitung oder Aufbewahrung von Körperzellen mit auf und spricht nicht mehr von Perfusion - wie dies in der RL der Fall ist - sondern von Infusion. Im zweiten Unterpunkt der Klassifizierungsregel 2 definiert die MDR nun auch die Aufbewahrung von Körperzellen und -geweben und weist Blutbeutel - so wie dies RL 93/42/EWG in Klassifizierungsregel 18 tut - direkt die Risikoklasse IIb zu.
- Regel 3 - Hier schließt die MDR nun auch die Veränderung der biologischen oder chemischen Zusammensetzung von menschlichen Geweben oder Zellen mit ein. Im Vergleich dazu, spricht die RL 93/42/EWG „nur“ von Veränderung des Blutes. Neu innerhalb dieser Regel der MDR ist auch, dass die Implantation oder auch die Verabreichung von Körperflüssigkeiten berücksichtigt wird. Ein im Vergleich zur RL vollkommen neuer Aspekt, der in diese Klassifizierungsregel Nr.3 mit einfließt, sind jene nicht invasiven Produkte, die aus einem Stoff oder einer Mischung aus Stoffen bestehen, die wiederum für den In-vitro-Gebrauch in unmittelbaren Kontakt mit dem menschlichen Körper entnommenen menschlichen Zellen, Geweben oder Organen oder für den In-vitro-Gebrauch mit menschlichen Embryonen vor derer Implantation oder Verabreichung in den Körper bestimmt sind. Diese nicht invasiven Produkte werden direkt der Risikoklasse III zugeordnet.
- Regel 4 - Die Regel 4 der MDR Klassifizierungsregeln unterscheidet sich von jenen der MDR dadurch, dass auch bei deren „Unterregeln“ die Schleimhäute explizit mitaufgenommen werden. Obwohl Regel 4 in der Produktkategorie der “Nicht invasiven Produkte” angeführt ist, gilt diese Regel laut MDR auch für invasive Produkte, die mit verletzter Schleimhaut in Berührung kommen.
Invasive Produkte: Die Klassifizierungsregeln der Produkt-Kategorie der “Invasiven Produkte” erfährt in der MDR vor allem bei der Anwendung der Regel 8 wesentliche Neuerungen.
- Regel 5 - gleichbleibend
- Regel 6 - gleichbleibend
- Regel 7 - Bei der die zur kurzzeitigen Anwendung bestimmten chirurgisch-invasiven Produkten betreffenden Klassifizierungsregel gibt beim Vergleich zwischen RL & MDR zwei Unterschiede. Zum einen gibt es in der MDR nun anstelle von fünf, sechs Unterregel. Dies rührt daher, dass Unterregel Nr.5 der RL thematisch aufgesplittet wurde in Produkte die im Körper eine chemische Änderung erfahren und Produkte zur Arzneimittelabgabe. Zum anderen gibt es in der fünften Unterregel der MDR bei den Produkten, die im Körper eine chemische Veränderung erfahren eine Erhöhung der Risikoklassen von IIb auf III.
- Regel 8 - Diese Klassifizierungsregel adressiert sowohl implantierbare Produkte, als auch zur langzeitigen Anwendung bestimmte chirurgisch-invasive Produkte. Vergleicht man RL & MDR, so gibt es bei der MDR nun neun, anstelle von fünf Unterregeln. Alle vier neu hinzukommenden Unterregeln spezifizieren Medizinprodukte, die der Medizinprodukte-Klasse III zuzuordnen sind:
- Aktiv implantierbare Produkte und dessen Zubehör
- Brustimplantate und chirurgische Netze
- Total- oder Teilprothesen von Gelenken (Zubehörkomponenten wie Schrauben, Keile, Platten und Instrumenten ausgenommen).
- Implantate zum Ersatz der Bandscheibe und implantierbare Produkte, die mit der Wirbelsäule in Berührung kommen (Zubehörkomponenten wie Schrauben, Keile, Platten und Instrumenten ausgenommen).
Aktive Produkte: Mit den Klassifizierungsregeln der Produkt-Kategorie “Aktive Produkte” wird in der MDR Software explizit einer eigenen Regel zugeordnet.
- Regel 9 - Im Vergleich zur RL, listet die MDR innerhalb dieser Klassifizierungsregel zwei zusätzlich Produkt-Gruppen mit auf. Zum einen definiert sie aktive Produkte, die zum Aussenden ionisierter Strahlung für therapeutische Zwecke bestimmt sind - inkl. jener Komponenten, die solche Produkte steuern und deren Leistung beeinflussen - als Medizinprodukte der Klasse IIb. Zum anderen adressiert die MDR in dieser Regel erstmals explizit Produkte, die dazu bestimmt sind, die Leistung von aktiven implantierbaren Produkten zu steuern, zu kontrollieren oder direkte zu beeinflussen und weist diesen die Risikoklasse III zu.
- Regel 10 - Bei der Regel für aktive Produkte mit Diagnose- und Überwachungszweck spricht die MDR im Vergleich zur RL in der ersten Unterregel nicht mehr von Absorption von Energie des menschlichen Körpers, sondern von Resorption. Des weiteren werden in dieser ersten Unterregel der MDR nun Produkten, die dazu bestimmt sind, den Körper des Patienten im sichtbaren Spektralbereich auszuleuchten direkt der Risikoklasse I zugeordnet. In der dritten Unterregel der Klassifizierungsregel 10 erweitert die MDR die Definition der direkten Diagnose oder Kontrolle um jene Diagnosen in klinischen Situationen, in denen der Patient in unmittelbarer Gefahr schwebt - IIb.
- Regel 11 - Mit der Klassifizierungsregel 11 in der MDR kommt es zu der offensichtlichsten Neuerung im Hinblick auf den Verglich zwischen RL 93/42/EWG und MDR. Die MDR geht bei dieser Regel explizit auf Software ein. Software, die dazu bestimmt ist, Informationen für diagnostische oder therapeutische Zwecke bereitszustellen werden der Risikoklasse IIa zugordnet. Sobald diese Informationen bzw. die daraus abgeleiteten Entscheidungen jedoch schwerwiegenden Einfluss auf die Gesundheit des Patienten (Tod oder irreversible Verschlechterung des Gesundheitszustandes) haben können, werden sie der höchsten Risikoklasse - III - zugeordnet. Kommt es aufgrund des Software-Einsatzes “nur” zu einer Verschlechterung (reversibel) des Gesundheitszustandes des Patienten, wird dem SW-Produkt die Risikoklasse IIb zugeordnet. Im letzten Absatz dieser Klassifizierungsregel definiert die MDR die für die Kontrolle von physiologischen Prozessen eingesetzte Software als Medizinprodukt der Klasse IIa - es sei denn, diese Kontrolle spezialisiert sich auf Vitalparameter, in diesem Fall kommt die Risikoklasse IIb zum Tragen. Alle anderen Software-Produkte / Software-Komponenten, die nicht in die oben angeführten Bestimmungen fallen, werden der Risikoklasse I zugeordnet.
- Regel 12 - Klassifizierungsregel 12 der MDR ist mit Regel 11 der RL gleichzusetzen.
- Regel 13 - Klassifizierungsregel 13 der MDR ist mit Regel 12 der RL gleichzusetzen.
Besondere Regeln: Die letzte Kategorie der Klassifizierungsregeln repräsentieren die sogenannten “Besonderen Regeln”. Vergleicht man RL 93/42/EWG und MDR, so gibt es in dieser Klassifizierungskategorie die größten Unterschiede.
- Regel 14 - Regel 14 der MDR ist mit Regel 13 der RL weitestgehend gleichzusetzen. Diese Regel weist Produkten, zu deren Bestandteilen ein Stoff gehört, der für sich alleine genommen als Arzneimittel zu definieren ist, die höchst Risikoklasse zu - III. Die MDR definiert diese Regel etwas ausführlicher und nimmt auch Arzneimittel aus menschlichem Blut oder Gewebe mit auf.
- Regel 15 - Regel 15 der MDR ist mit Regel 14 der RL gleichzusetzen.
- Regel 16 - Regel 16 der MDR ist Großteils mit Regel 15 der RL zu vergleichen. Die MDR nimmt bei den Produkten, die speziell zum Desinfizieren von Medizinprodukten vorgesehen sind, auch die Sterilisation mit auf. Desinfektionslösungen oder auch Reinigungs-Desinfektionsgeräte schließt die MDR aus dieser Regel explizit aus.
- Regel 17 - Regel 17 der MDR ist mit Regel 16 der RL gleichzusetzen.
- Regel 18 - Diese Regel der MDR ist nur mehr in sehr geringem Ausmaß mit Regel 17 der RL 93/42/EWG gleichzusetzen. Die MDR nimmt in diese Regel nun auch nicht lebensfähige oder abgetötete Gewebe oder Zellen sowohl menschlichen, als auch tierischen Ursprungs mit auf.
Die nun folgenden Klassifizierungsregeln - 19-22 - sind nur mehr in der MDR zu finden. Die RL 93/42/EWG beendet ihre Klassifizierungsregeln mit Regel 18 und der Definition, dass Blutbeutel der Klasse IIb zuzuordnen sind.
- Regel 19 - Diese Regel dient der Klassifizierung von Produkten, die Nanomaterial enthalten oder daraus bestehen. Die Zuordnung der Risikoklassen geht hierbei von IIa - III, basierend auf dem Schweregrad der möglichen internen Exposition der Nanomaterialien.
- Regel 20 - Regel 20 der MDR definiert invasive Produkte im Zusammenhang mit Körperöffnungen - außer chirurgisch invasiven Produkten - die für die Verabreichung von Arzneimitteln durch Inhalation bestimmt sind als Medizinprodukte der Klasse IIa - außer im Behandlungsfall lebensbedrohlicher Umstände, in diesem Fall werden sie der Klasse IIb zugeordnet.
- Regel 21 - Regel 21 der MDR klassifiziert Produkte, die aus Stoffen oder Kombinationen von Stoffen bestehen, die dazu bestimmt sind durch eine Körperöffnung in den menschlichen Körper eingeführt oder auf die Haut aufgetragen zu werden, und die vom Körper aufgenommen oder lokal im Körper verteilt werden. In diesem Fall werden diese Produkte den Risikoklassen IIa - III zugeordnet, anhängig von anatomischer Lokalisation des bestimmungsgemäßen Gebrauches.
- Regel 22 - Die letzte der Klassifizierungsregeln dient der Klassifizierung von aktiven therapeutischen Produkten mit integrierter oder eigebauter diagnostischer Funktion, die das Patientenmanagement bestimmen - wie etwa geschlossene Regelsysteme oder automatische externe Defibrillatoren.
Das gesamte RnB-Team möchte sich an dieser Stelle bei Herrn Armin Gärtner bedanken. Herr Gärtner hat uns netterweise auf die richtlinienkonforme Schreibweise der “CE-Kennzeichnung” aufmerksam gemacht. “CE-Kennzeichen” oder “CE-Zeichen” - wie urspünglich in diesem Blog-Artikel definiert - entspricht nicht der korrekten Definition laut RL 93/42/EWG und MDR.
MDR Teil 3 - Klassifizierungsregeln Rahmenbedingungen für die Anwendung - MDR & RL 93/42/EWG im Vergleich
Die letzten beiden Blog-Artikel diskutierten die grundlegenden Sicherheits- und Leistungsmerkmale der MDR. Dieser Beitrag geht nun auf die Rahmenbedingungen der Anwendung der Klassifizierungsregeln ein und stellt MDR & RL 93/42/EWG gegenüber. In der Medizinprodukte-Verordnung, sind die Klassifizierungsregeln in Anhang VIII zu finden - RL 93/42/EWG definiert die Klassifizierung von Medizinprodukten in Anhang IX.
Dauer & Art der Anwendung
Um dem Medizinprodukt eine Risikoklasse zuordnen zu können, müssen - vor Anwendung der Klassifizierungsregeln - produktspezifische Rahmenbedingungen festgelegt werden.
Die Dauer und Art der Anwendung spielen in diesem Zusammenhang eine wesentliche Rolle.

Dauer der Anwendung: Die Definitionen “Vorübergehend” - “Kurzzeitig” und “Langzeitig”, spezifizieren die Art der Dauer der Anwendung eines Medizinproduktes. Vergleicht man diese Definitionen von MDR und RL, so gibt es keine nennenswerten Änderungen, lediglich die Zeitspanne einer “kurzzeitigen” Anwendung wird in der MDR mit der Angabe von 60 Minuten bis 30 Tage exakter definiert.
Art der Anwendung: Bei der Art der Anwendung eines Medizinproduktes gibt es im Vergleich von MDR & RL einige nennenswerte Unterschiede. Die MDR nimmt bei der Definition eines “Chirurgisch-invasiven Produktes” nun auch “Schleimhäute der Körperöffnungen” mit auf, geht bei der Definition von “Wiederverwendbaren chirurgischen Instrumenten” auf geeignete Verfahren wie Reinigung, Desinfektion und Sterilisation ein und stellt klar, dass wiederverwendbare chirurgische Instrumente auch klar vom Hersteller als “wiederverwendbar” definiert sein müssen.
Die allgemeine Definition zu “Aktiven Medizinprodukten” der RL (Annex IX) ist in der MDR (Annex VIII) nicht mehr zu finden. Die MDR definiert - für die Rahmenbedingung der Klassifizierung - “aktive therapeutische Produkte” und “aktive Medizinprodukte zu Diagnose- und Überwachungszwecken”. Neu ist hier, dass Überwachungszwecke explizit in die Definition mitaufgenommen wurden. In den allgemeinen Begriffsbestimmungen der MDR (Annex II), wird nicht das aktive Medizinprodukt, sondern das “aktive Produkt” definiert. Diese Definition fehlt bei den Begriffsbestimmungen der RL 93/42/EWG Artikel 1 zur Gänze, ist jedoch mit der Definition “aktives Medizinprodukt” aus RL 93/42/EWG Annex IX zu vergleichen.
Mit der Definition “Verletzte Haut oder Schleimhaut” definiert die MDR - im Vergleich zur RL - eine vollkommen neue “Art der Anwendung” eines Medizinproduktes.
Durchführungsvorschriften / Anwendungsregeln
Für die Anwendung der Klassifizierungsregeln gelten sowohl in der RL (“Anwendungsregeln”), als auch in der MDR (“Durchführungsregeln”) gewisse Regeln, an die es sich zu halten gilt. Es ist unumstritten, dass sich die Anwendung der Klassifizierungsregeln nach der Zweckbestimmung des Produktes richtet. Wird das Medizinprodukt - laut seiner Zweckbestimmung - mit anderen Produkten verwendet, so sind die Klassifizierungsregeln auf jedes Produkt gesondert anzuwenden - selbiges gibt für Zubehör. Die MDR definiert in Anhang XVI ein Verzeichnis der Gruppen von Produkten ohne medizinischen Verwendungszweck. Wird das zu klassifizierende Medizinprodukt - laut seiner Zweckbestimmung - mit einem dieser Produkte zusammen verwendet, müssen diese gesondert klassifiziert werden.
 Für die Klassifizierung von Software ist - vergleicht man RL und MDR - gleichbleibend, dass Software, die ein Produkt steuert bzw. dessen Anwendung beeinflusst derselben Risikoklasse zugeordnet wird wie das Produkt. Neu ist, dass die MDR explizit darauf hinweist, dass Stand-alone Software produktunabhängig zu klassifizieren ist.
Für die Klassifizierung von Software ist - vergleicht man RL und MDR - gleichbleibend, dass Software, die ein Produkt steuert bzw. dessen Anwendung beeinflusst derselben Risikoklasse zugeordnet wird wie das Produkt. Neu ist, dass die MDR explizit darauf hinweist, dass Stand-alone Software produktunabhängig zu klassifizieren ist.
Auch die “Worst-Case-Scenario”-Betrachtung ist - vergleicht man RL und MDR - gleichgeblieben. Sobald mehrere Klassifzierungsregeln oder auch Unterregeln auf ein Produkt zutreffen, ist jene Regel mit der höchsten Risikoklasse anzuwenden.
Neu ist, dass die Auswahlkriterien der jeweiligen Klassifizierungsregel laut MDR Anhang II in der technischen Dokumentation des Medizinproduktes angeführt werden müssen. Die Regeln müssen iterativ bewertet und Auschluss- und letztenendes Auswahlkriterien dokumentiert werden.
Ausblick: Der nächste Blog-Artikel wird die Klassifizierungsregeln von MDR und RL 93/42/EWG gegenüberstellen.
MDR Teil 2 - Überblick über die grundlegenden Sicherheits- und Leistungsanforderungen - Allgemeine Anforderungen
Der letzte Blog-Artikel zur MDR stellte die grundlegenden Sicherheits-und Leistungsanforderungen als Überblick dar. Dieser Beitrag soll nun weiterführend auf die erste Kategorie dieser Anforderungen eingehen: Die allgemeinen Anforderungen an Medizinprodukte.
An der grundsätzlichen Zielausrichtung aus regulatorischer Sicht hat sich nichts geändert - es müssen sowohl die Sicherheit als auch die Wirksamkeit ermittelt und in ein Verhältnis gebracht werden. Dieses Verhältnis muss gemäß dem definierten klinischen Kontext akzeptabel sein. Zusätzlich muss Vertrauen geschaffen werden, dass sich diese beiden Komponenten während der Lebensdauer des Medizinprodukts nicht maßgeblich verändern (Zuverlässigkeit) und die vorhersehbare Fehlbedienung in die Erstellung dieses Verhältnisses mit einfließt.

Die entsprechenden Leistungsangaben beziehen sich hierbei auf die Angaben des Herstellers – entsprechende Daten zur klinischen Wirksamkeit und der Sicherheit sollten also in den Begleitinformationen des Produkts zu finden sein (gestützt mit Evidenz aus der klinischen Bewertung und zusätzlichen Validierungstätigkeiten).
Gefordert wird, wenig überraschend, ein systematischer Risikomanagementprozess. Dezidiert erwähnt ist hierbei dessen iterativer Charakter und die fortlaufende Aktualisierung auch nach der Markteinführung. Die Reihenfolge der Risikokontrollmaßnahmen ist ebenfalls definiert – inhärente Sicherheit durch Auslegung und Herstellung, Schutzmaßnahmen (Alarmvorrichtungen) und Sicherheitsinformationen (Begleitinformationen) stellen das bereits bekannte Stufensystem der Kontrollmaßnahmen dar.
Der Bezug zum gebrauchstauglichkeitsorientierten Entwicklungsprozess wird ebenfalls erneut klar beschrieben. Dies äußert sich unter anderem durch die Anforderungen, welche das systematische Management von Risiken aufgrund von ergonomischen Merkmalen, Umgebungsbedingungen sowie aufgrund von speziellen Charakteristika und Kenntnissen von Patienten und Anwender erfordern. Hierbei sei erwähnt, dass der „normale Gebrauch“ im Kontext der Norm IEC 62366 neben dem korrekten Gebrauch auch vorhersehbare Benutzungsfehler einschließt.
Grundsätzlich sind diese allgemeinen Prozesse des Risikomanagements, der gebrauchstauglichkeitsorientierten Entwicklung und der klinischen Bewertung bereits aus der RL 93/42/EWG bekannt.
Unterschiede zu den allgemeinen Anforderungen des Anhangs I der RL 93/42/EWG:
- der Risikomanagementprozess wird detaillierter beschrieben, bildet aber im Wesentlichen den Prozess gemäß ISO 14971 ab
- Risiken müssen soweit verringert werden, wie es ohne negative Auswirkung auf das Nutzen/Risiko-Verhältnis möglich ist
- die Anforderung einer klinischen Bewertung ist nicht dezidiert als Anforderung gelistet, findet sich aber implizit in anderen wieder
- der iterative Charakter des Risikomanagements inklusive Marktüberwachung wird deutlicher erwähnt
- für Produkte ohne medizinische Zweckbestimmung (Anhang XVI – Kontaktlinsen für kosmetische Zwecke…) liegt der Fokus auf den Sicherheitsaspekten
Erfahren Sie im nächsten Artikel mehr über die Klassifizierung von Medizinprodukten in der MDR.
"Mach es fertig - bevor es dich fertig macht"
Zulassung von Medizinprodukten: RnB-Consulting steht für ganzheitliche Projektabwicklung & Betreuung

Uns als Beratungsunternehmen ist es ein persönliches Anliegen, Medizinprodukte-Herstellern eine ganzheitliche Projektabwicklung anbieten zu können. Wir stehen als zentrale Anlaufstelle für Fragen zu den unterschiedlichsten Themenbereichen, die es im Laufe eines Zulassungsprozesses zu analysieren und zu bearbeiten gilt, zur Verfügung - Gebrauchstauglichkeit, Risikomanagement, Klinische Bewertung, Elektrische Sicherheit, Biokompatibilität, Software-Dokumentation (um nur einige wenige zu nennen).

Damit Sie als Medizinprodukte-Hersteller aufgrund der Vielzahl an Disziplinen, die es für die Zulassung eines Medizinproduktes zu bewältigen gilt, nicht verzweifeln, stehen wir Ihnen mit unserer Expertise zur Verfügung. Die initiale Phase besteht darin, gemeinsam Firmen- und Produktinfos zusammenzuführen. Ziel ist, Ihnen auf Basis der gesammelten Daten eine GAP-Analyse der technischen Dokumentation und so einen repräsentativen Kostenvoranschlag - inkl. aller notwendigen Arbeitspakte - zur Verfügung stellen zu können. Nach Ihrer Auftragserteilung erhalten Sie einen detaillierten Projektmanagement-Plan inkl. aller notwendigen Arbeitspakete. Ein wesentlicher Schritt ist hierbei das persönliche Kennenlernen aller RnB-Kooperationspartner. Zusammen erarbeiten wir Anforderungen der oben bereits angesprochenen Disziplinen wie Biokompatibilität, Gebrauchstauglichkeit, uvm.
Nun geht es an die kontrollierte Abarbeitung aller Arbeitspakete. Wöchtenliche Projekt-Statusberichte ermöglichen es Ihnen, den Überblick zu behalten und anstehende Arbeitsschritte proaktiv zu organisieren. Persönliche Treffen zwischen Ihnen und uns halten das Projekt am Laufen und sorgen für klare Projektstrukturen.
Gerne übernehmen wir auch für Sie die Kontaktaufnahme zu Benannten Stellen, um Angebote zur Bewertung der Konformität zu erhalten.

Parallel zur Begleitung von Zertifizierungsprojekten bieten wir auch ein breites Spektrum an Schulungen an. Um Sie und Ihre Mitarbeiter effektiv auf das akutelle regulatorische Rahmenwerk der Medizintechnik zu schulen, erhalten Sie durch Praxisbeispiele einen guten Einblick in die verschiedensten Möglichkeiten der Erfüllung regulatorischer Gegebenheiten. Eine Wissensüberprüfung inkl. Zertifikat bei erfolgreicher Absolvierung runden unser Angebot ab.