Vigilanzsystem gemäß (EU) 2017/745
Meldepflicht gemäß der MDR
Wird ein Vorkommnis nach der ersten Bewertung nicht als schwerwiegendes Vorkommnis eingestuft, muss dennoch geprüft werden, ob es zu einem der in Artikel 2 (65) a-c der MDR genannten Ergebnisse führen könnte, wenn die Umstände weniger günstig wären (z. B. ohne die Intervention eines Dritten oder wenn mehr gefährdete Patienten derselben Situation ausgesetzt gewesen wären usw.).
Wenn der Hersteller nicht ausschließen kann, dass das Vorkommnis möglicherweise zu den in Artikel 2 (65) a- c MDR genannten Folgen geführt haben könnte, ist das Vorkommnis als schwerwiegend zu betrachten und muss der zuständigen Behörde gemeldet werden. Ist sich der Hersteller nach Bekanntwerden eines möglicherweise meldepflichtigen Vorkommnisses unsicher darüber, ob das Vorkommnis meldepflichtig ist, muss er dennoch innerhalb der in Artikel 87(2) gemäß Artikel 87 (2-5) MDR einen Bericht vorlegen.
Das nachstehende Flussdiagramm veranschaulicht den von den Herstellern zu befolgenden Prozess für das Management von Vorkommnissen und schwerwiegenden Vorkommnissen.
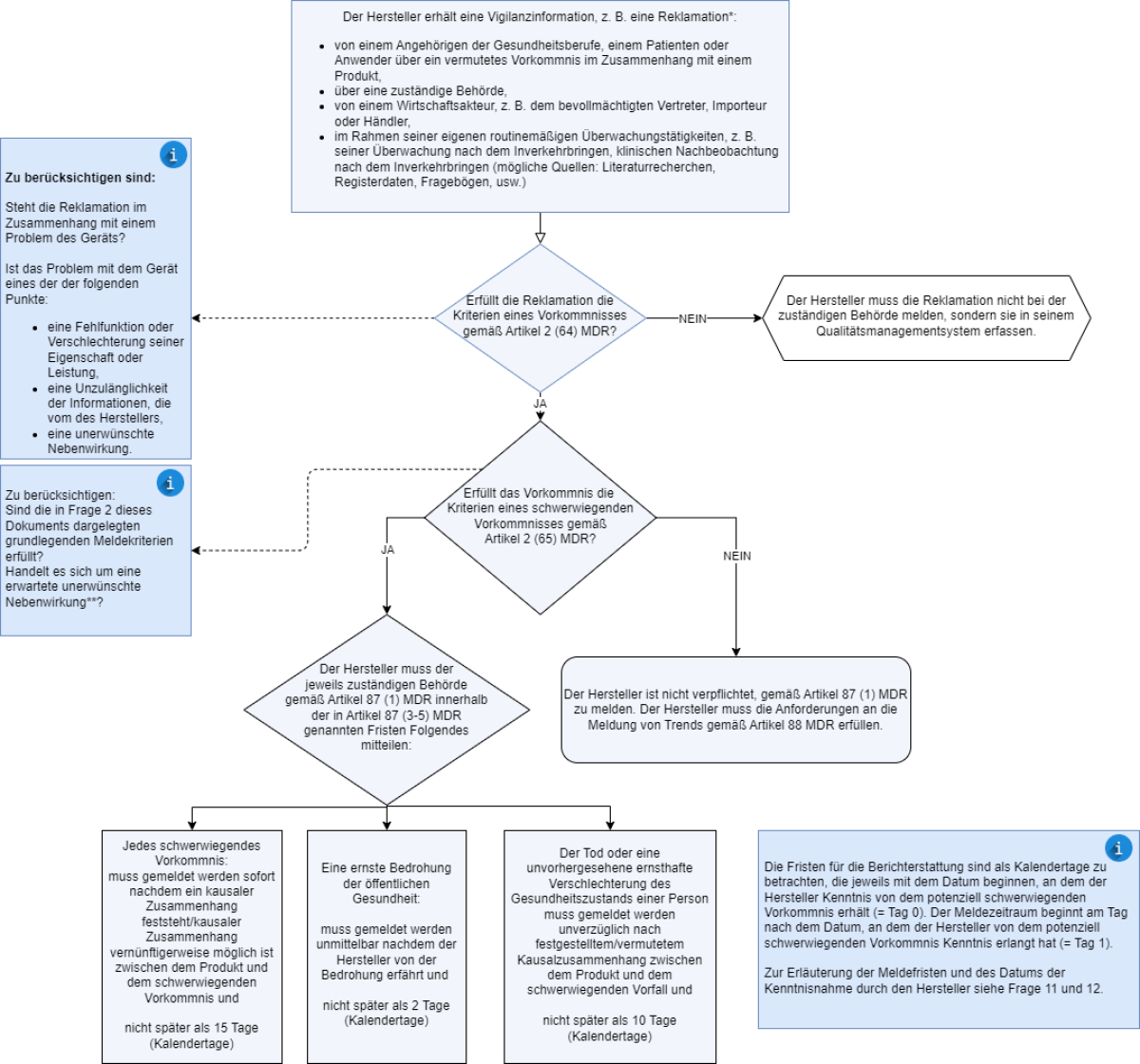
Bilderquelle: Leitfaden MDCG 2023-3 “Questions and Answers on vigilance terms and concepts as outlined in the Regulation (EU) 2017/745 on medical devices”, Seite 4.
Was versteht man unter einer Reklamation?
Eine Reklamation ist in der EN ISO 13485:2016 definiert und kann als schriftliche, elektronische oder mündliche Mitteilung beschrieben werden, die sich auf angebliche Unzulänglichkeiten in Bezug auf die Identität, Qualität, Haltbarkeit, Zuverlässigkeit, Gebrauchstauglichkeit, Sicherheit oder Leistung eines Medizinprodukts oder im Zusammenhang mit einer Dienstleistung, die die Leistung eines solchen Medizinprodukts beeinträchtigt. Es ist wichtig zu beachten, dass Reklamationen und Informationen, die für die Vigilanzmeldung relevant sind, nicht nur von außen kommen, sondern auch aufgrund der eigenen Aktivitäten des Herstellers zur routinemäßigen Überwachung der Sicherheit und Leistung eines Produkts entstehen können.
Was versteht man unter unerwünschte Nebenwirkungen?
Unter einer “unerwünschten Nebenwirkung” im Sinne der MDR ist jede unbeabsichtigte und unerwünschte medizinische Erscheinung im menschlichen Körper, die sich aus der normalen Anwendung eines Produkts ergibt. Unerwünschte Nebenwirkungen sind nicht das Ergebnis einer Fehlfunktion, einer Verschlechterung der Eigenschaften oder der Leistung des Produkts oder einer unzureichenden Information durch den Hersteller. Eine erfolglose Behandlung (oder ein Behandlungsfehler) sollte nicht als unerwünschte Nebenwirkung angesehen werden. Für die Zwecke dieses Leitfadens können unerwünschte Nebenwirkungen erwartet oder unerwartet sein und werden als Zwischenfälle im Sinne der MDR betrachtet (Artikel 2(64) MDR).
Erwartete unerwünschte Nebenwirkungen werden in der Produktinformation klar dokumentiert, in der technischen Dokumentation quantifiziert und unterliegen der Meldung von Trends gemäß Artikel 88 MDR.
Der Unterschied zwischen Vorkommnis und schwerwiegendes Vorkommnis
„Vorkommnis“ bezeichnet eine Fehlfunktion oder Verschlechterung der Eigenschaften oder Leistung eines bereits auf dem Markt bereitgestellten Produkts, einschließlich Anwendungsfehlern aufgrund ergonomischer Merkmale, sowie eine Unzulänglichkeit der vom Hersteller bereitgestellten Informationen oder eine unerwünschte Nebenwirkung.
„Schwerwiegendes Vorkommnis“ bezeichnet ein Vorkommnis, das direkt oder indirekt eine der nachstehenden Folgen hatte, hätte haben können oder haben könnte:
- den Tod eines Patienten, Anwenders oder einer anderen Person
- die vorübergehende oder dauerhafte schwerwiegende Verschlechterung des Gesundheitszustands eines Patienten, Anwenders oder anderer Personen
- eine schwerwiegende Gefahr für die öffentliche Gesundheit
Nähere Informationen zu Begriffen und Konzepte der Vigilanz finden Sie im folgenden MDCG Leitfaden: Leitfaden MDCG 2023-3