Blog
Die MDR hat vollständige Gültigkeit erhalten - wir geben einen Überblick
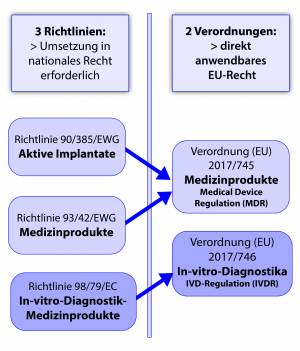 Durch die Umstellung auf die neue MDR/IVDR verschmelzen 3 Richtlinien zu zwei Verordnungen. Diese Verordnungen wirken als direkt anwendbares EU-Recht. Die MDR ist seit 26.05.21 bereits gültig und der Geltungsbeginn der IVDR für den 26.05.2022 festgelegt.
Durch die Umstellung auf die neue MDR/IVDR verschmelzen 3 Richtlinien zu zwei Verordnungen. Diese Verordnungen wirken als direkt anwendbares EU-Recht. Die MDR ist seit 26.05.21 bereits gültig und der Geltungsbeginn der IVDR für den 26.05.2022 festgelegt.
Was ist neu?
Es werden wesentliche Schlüsselelemente gestärkt und eine bessere Transparenz und Rückverfolgbarkeit gewährleistet:
1) Erhebliche Stärkung von Schlüsselelemente
- Beaufsichtigung der benannten Stellen
- Risikoklassifizierung
- Konformitätsbewertungsverfahren
- Klinischer Bewertung
- Vigilianz und Marktüberwachung
2) Gewährleistung von Transparenz und Rückverfolgbarkeit in Bezug auf Medizinprodukte
- Einmalige Produkterkennung (UDI-Unique Device Identification)
- Einmalige Registrierungsnummer (SRN – Single Registration Number)
Im Detail beinhaltet die MDR folgende Anforderungen:
- Der Anwendungsbereich hat sich deutlich vergrößert und umfasst auch Produkte zur Reinigung, Sterilisation oder Desinfektion anderer Medizinprodukte und spezielle Produkte ohne medizinischen Zweck
- Der Sicherheitsaspekt orientiert sich am gesamten Produktlebenszyklus und wird durch klinische Daten verdeutlicht
- Es werden höhere Anforderungen an benannte Stellen geltend
- Es wird ein unabhängiges Expertengremium bei der klinischen Bewertung von Hochrisiko-Produkten eingeführt
- Ein System für bessere Marktüberwachung und Rückverfolgung wurde eingeführt (UDI, EUDAMED)
- Für Hersteller ist eine Eingabe von Daten bei EUDAMED erforderlich
Anforderungen an Hersteller
Die MDR verpflichtet Hersteller in Artikel 10:
- zu einem Risikomanagementsystem
- zu einem Qualitätsmanagementsystem
- zu einer klinische Bewertung
- zu einer technische Dokumentation
- sowie ein Konformitätsbewertungsverfahren anzuwenden
Der Hersteller muss für das Inverkehrbringen und die Inbetriebnahme seiner Produkte gewährleisten, dass diese gemäß den Anforderungen der europäischen Verordnung ausgelegt und hergestellt sind. 1
Hinsichtlich der Forderung eines QM Systems wird in Artikel 10 MDR folgendes festgehalten:
„Die Hersteller von Produkten, bei denen es sich nicht um Prüfprodukte handelt, müssen ein Qualitätsmanagementsystem einrichten, dokumentieren, anwenden, aufrechterhalten, ständig aktualisieren und kontinuierlich verbessern, da die Einhaltung dieser Verordnung auf die wirksamste Weise sowie einer der Risikoklasse und der Art des Produkts angemessenen Weise gewährleistet.“
Zusammenhang MDR & Harmonisierte Normen
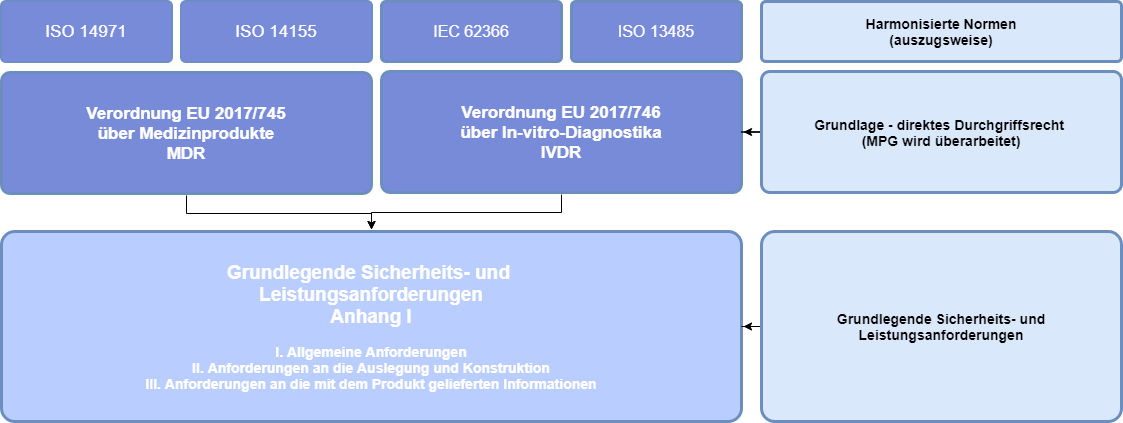
Zentrales Element des New Approach im Rahmen der Binnenmarktregelungen sind die in Anhang I (MDR) dargelegten „Grundlegende Sicherheits- und Leistungsanforderungen“ (in der MDD noch als Grundlegende Anforderungen bezeichnet). Medizinprodukte müssen die unter Berücksichtigung ihrer Zweckbestimmung für sie anwendbaren grundlegenden Anforderungen erfüllen, damit sie in der EU in Verkehr gebracht/ in Betrieb genommen werden können1 (siehe Abbildung unten):
- Kap. 1 Allgemeine Anforderungen
- Kap. 2 Anforderungen an Auslegung und Herstellung
- Kap. 3 Anforderungen an die mit dem Produkt gelieferten Informationen (Kennzeichnung, Gebrauchsanweisung, Verpackung)
Für den Nachweis der Grundlegenden Sicherheits- und Leistungsanforderungen (GSLA) müssen die Hersteller, die angewendeten harmonisierten Normen in der GSLA referenzieren, um somit die Konformität des Produktes darzulegen. Sehr wesentliche Normen sind hierbei unter anderem ISO 14971 (Risikomanagement), ISO 14155 (Gute klinische Praxis), IEC 62366 (Gebrauchstauglichkeit) und die ISO 13485 (Qualitätsmanagement).
Hilfestellung finden Hersteller in folgenden Dokumenten oder auf folgenden Websiten:
- MDR - Link zur MDR
- IVDR - Link zur IVDR
- EUDAMED - EUDAMED
- GS1 - GS1
- IMDRF - IMDRF
- MEDDEV - MEDDEV Guidance
- MDCG Dokumente -MDCG
1 Buch: Medizinprodukte und IVD - Fit für Europa -Medizinprodukte und IVD - Fit für Europa
Es war einmal die MDD...
 Mit heutigem Datum 26.05.21 erlangt die MDR ihre vollständige Gültigkeit. Medizinprodukte müssen nach MDR zugelassen werden, um In-verkehr gebracht werden zu dürfen. Medizinprodukte die eine MDD Zertifizierung vor dem 26. Mai 2021 erhalten haben, können bis zum 26. Mai 2025 auf den Markt gebracht werden, sofern diese Zertifikate nicht früher ablaufen. Danach muss jedes Medizinprodukt nach MDR zertifiziert sein.
Mit heutigem Datum 26.05.21 erlangt die MDR ihre vollständige Gültigkeit. Medizinprodukte müssen nach MDR zugelassen werden, um In-verkehr gebracht werden zu dürfen. Medizinprodukte die eine MDD Zertifizierung vor dem 26. Mai 2021 erhalten haben, können bis zum 26. Mai 2025 auf den Markt gebracht werden, sofern diese Zertifikate nicht früher ablaufen. Danach muss jedes Medizinprodukt nach MDR zertifiziert sein.
Welche Konsequenzen trägt die Änderung mit sich?
- Änderungen in der Klassifizierung
- Höhere Sicherheits- und Leistungsanforderungen an Produkte
- Wesentlich höhere Anforderungen an die Überwachung nach dem Inverkehrbringen
- Höhere und detailliertere Anforderungen an die technische Dokumentation
- Kürzere Meldefristen bei der Meldung von schwerwiegenden Vorkommnissen
- Zusätzliche Anforderungen an das Qualitätsmanagementsystem
- Einführung einer UDI-Kennzeichnung
- Änderungen im OEM-PLM Verhältnis
- Strengere Anforderungen an benannte Stellen
- Höhere Anforderung an die Klinische Bewertung
Buchempfehlung:
Expert*innen der Neurochirurgie dienen als Ideengeber für perfektes Design von MEDUSA.
In den vergangenen Monaten haben zwischen dem Usability-Team des Leitprojektes „MEDUSA“ und Neurochirurg*innen des Kepler Universitätsklinikums bzw. des Neuromed Campus sogenannte Experten-Interview stattgefunden.

Ziel dieser Interviews war es, ein aussagekräftiges Feedback zum geplanten ersten Prototyp zu erhalten. Das Sammeln von Ideen und vor allem Wünschen an die künftige neurochirurgische Trainingsplattform hatte dabei oberste Priorität. Denn für das gesamte Projektteam ist es wichtig, bereits in früher Projektphase zu erfahren, welche Funktionalitäten und vor allem Designs in Hinblick auf den hybriden Ansatz* und der Trainingsplattform-Umsetzung seitens der Expert*innen als relevant und welche als nicht relevant angesehen werden.
Funktionelle Umsetzungen und Designs, die im Zuge der Interviews als nicht relevant eingestuft wurden, repräsentieren aktuell den wohl wichtigsten Input, um den weiteren Projektverlauf gezielt planen und Prototypen – „in time“ – realisieren zu können. Der nächste Schritt wird – sofern es die Auflagen aufgrund der aktuellen Situation zulassen – das projektinterne Testen des ersten Prototyps in den Räumlichkeiten der MEDUSA-Garage sein. Anhand dieses projektinternen Testens werden letzte Änderungen am allerersten Prototyp vorgenommen werden.
Dieser erste Prototyp wird selbstverständlich von Expert*innen der Neurochirurgie getestet werden. Die Ergebnisse dieser Testdurchläufe werden wiederum als Design-Input für Hard- und Software-Entwicklung für die Umsetzung des zweiten Prototyps dienen.
Auf diesem Wege möchte sich das gesamte MEDUSA-Projektteam bei den Teilnehmer*innen der Interviews und den damit gelieferten wichtigen Inputs recht herzlich bedanken!
* Kombination von realen und virtuellen Komponenten
Registrierungspflichten - UKCA Kennzeichnung in Großbritannien

Medizinprodukte und IVD müssen seit dem 01.01.2021 nach den neuen Registrierungs- und Kennzeichnungsrichtlinien in Großbritannien in Verkehr gebracht werden.
Für den Marktzugang in Großbritannien ist die UKCA-Kennzeichnung entwickelt worden. Das bisherige CE-Kennzeichen findet somit bald keine Anerkennung auf diesem Markt mehr und wird durch das UKCA Zeichen ersetzt.1
Seit dem 1. Januar 2021 müssen alle Medizinprodukte, einschließlich In-vitro-Diagnostika (IVDs), die in Großbritannien auf den Markt gebracht werden, bei der MHRA (The Medicines and Healthcare products Regulatory Agency) registriert werden.
Die Schonfristen der MHRA Registrierung/Umstellungspflicht sind abhängig von der Klassifizierung:
- Für implantierbare Produkte der Klasse III und Klasse IIb, alle aktiven implantierbaren Medizinprodukte und Produkte der IVD-Liste A gilt die Umstellungspflicht der Kennzeichnung ab 1. Mai 2021
- Andere Produkte der Klasse IIb und alle Produkte der Klasse IIa sowie Produkte der IVD-Liste B und IVDs zur Eigenanwendung unterliegen ab dem 1. September 2021 der neuen Kennzeichnungspflicht
- Klasse I und sonstige IVDs müssen ab 1. Januar 2022 registriert werden
Weitere Informationen zur Registrierung (einschließlich Gebühren) finden Sie unter folgendem Link: MHRA Registrierung.2
Auch britische Produkte haben keinen Zugang mehr zum europäischen Markt, denn seit Jänner 2021 sind benannte Stellen aus UK in der EU nicht mehr akkreditiert. Dies hat zur Folge, dass UK Produkte für den europäischen Markt eine Konformitätsbewertung einer benannten Stelle in der EU benötigen.3
Für Irland reicht auch das UKCA Kennzeichen nicht aus, hier wird die CE- oder UKNI Kennzeichnung benötigt.
Zusammenfassung der wichtigsten Anforderungen für das Inverkehrbringen in Großbritannien:
- CE-Kennzeichnung und Zertifikate von benannten Stellen mit Sitz in der EU bleiben bis 30. Juni 2023 in Großbritannien anerkannt.
- Die EU erkennt die benannten Stellen des Vereinigten Königreichs nicht mehr an
- Benannte Stellen im Vereinigten Königreich sind nicht in der Lage, CE-Zertifikate auszustellen (ausgenommen “CE UKNI”-Kennzeichnung - gültig in Nordirland)
- Seit 1. Jänner 2021 müssen alle Medizinprodukte bei der zuständigen Behörde “Medicines and Healthcare products Regulatory Agency (MHRA)” registriert werden. Die Fristen zur Registrierung betragen vier bis zwölf Monate.
- Hersteller mit Sitz außerhalb Großbritanniens müssen einen verantwortlichen Repräsentanten (UK Responsible Person) benennen.
1 devicemed.de
2 gov.uk
3 diapharm.com
MEDUSA in den Fußstapfen von großen Technikfirmen im Linzer Silicon Valley

In den Fußstapfen von großen Technikfirmen, wie Microsoft, Amazon, Google und Apple, die ihren Erfolg unter anderem einer Garage widmen können, wurde nun die MEDUSA-Garage im Linzer Silicon Valley initialisiert.
In der MEDUSA-Garage, werden nun die innovativen Ideen, die seit 2019 konzipiert wurden, verwirklicht. Je größer ein Projekt ist, desto ausgeprägter sind die einzelnen Funktionen. Dadurch wird es umso schwieriger das Gesamtkonzept und seine Auswirkungen zu erkennen. Vor allem deswegen ist es nun essenziell die einzelnen konzipierten Komponenten und Expertisen aller Konsortialpartner in der MEDUSA-Garage zu vereinen, denn „Das Ganze ist mehr als die Summe seiner Teile“ – Aristoteles.
Am 22.09.2020 fand bereits die FFG-Zwischenevaluierung statt, bei der die 13 Konsortialpartnerinnen und Konsortialpartner mit der Präsentation des Projektfortschritts und geplanten Vorgehensweisen glänzen konnten. Die positive Bewertung und das konstruktive Feedback durch die Fachevaluatorinnen und Fachevaluatoren sind Antrieb und Bestätigung für das ganze Konsortium.
Das Ziel des Forschungsprojekts MEDUSA ist die Entwicklung einer revolutionären Trainings- und Planungsplattform für Neurochirurginnen und Neurochirurgen, um komplexe Clipping-Operationen an Gehirnarterien detailreich und ganzheitlich simulieren zu können. Ein hybrider neurochirurgischer Simulator soll die reale und virtuelle Welt miteinander verbinden, um vielseitige und realistische Trainingsmöglichkeiten zu schaffen. Chirurginnen und Chirurgen können so die künstlich gefertigte Patientin oder den künstlich gefertigten Patienten haptisch fühlen und innere, ansonsten nicht sichtbare anatomische Strukturen in Form von virtuell erzeugten Hologrammen sehen.
Projektpartner: alpha medical concepts e.U, cortEXplore GmbH, eulerian-solutions e.U, EVO-tech GmbH, FH OÖ Forschungs- und Entwicklungs-GmbH, Johannes Kepler-Universität-Institute of Polymer Product Engineering, Johannes Kepler-Universität - Institut für Polymerwissenschaften, Kepleruniversitätsklinikum Linz - Universitätsklinik für Neurochirurgie, LIFEtool gemeinnützige GmbH, Netural GmbH, RISC Software GmbH, R'n'B Consulting GmbH, Profactor GmbH
Nähere Informationen zum Projekt finden Sie unter: MEDUSA WEBSITE