Blog
Ablösung der EN1041 und ISO 15223-1:2017 durch ISO 20417:2021 und ISO15223-1:2021

Die Normen werden ohne EU-Harmonisierung obligatorisch sein und werden im ersten Quartal 2021 erwartet.
In der ISO 15223-1:2021 wurden die Symbole und Definitionen geprüft. Weiters wurden im bereits vorliegenden Entwurf ISO/DIS 15223-1:2020-04 ca. 20 neue Symbole ergänzt.
Neu ist bei der ISO20417:2021 auch das MD (Medical Device) Zeichen, welches zukünftig auf jedem Medizinprodukt und/oder Verpackung angebracht werden muss, um das Medizinprodukt erkenntlich zu machen. Produkte, bei denen nach einer ausführlichen Risikobetrachtung keine Kennzeichnung angebracht werden kann oder bei denen die Kennzeichnung Risiken hervorrufen oder steigern, sind von dieser Pflicht ausgenommen (Kapitel 4.2 ISO/DIS 20417).
Weiters müssen Medizinprodukte, die ihre Konformität durch die britische Behörde erlangt haben, das UKCA Kennzeichen (United Kingdom Conformity Assessment) tragen.
RnB-Consulting unterstützt Sie im Rahmen der Implementierung der europäischen Verordnungen für Medizinprodukte und In vitro Diagnostika (MDR und IVDR) und steht dabei mit erfahrenen Experten zur Seite.
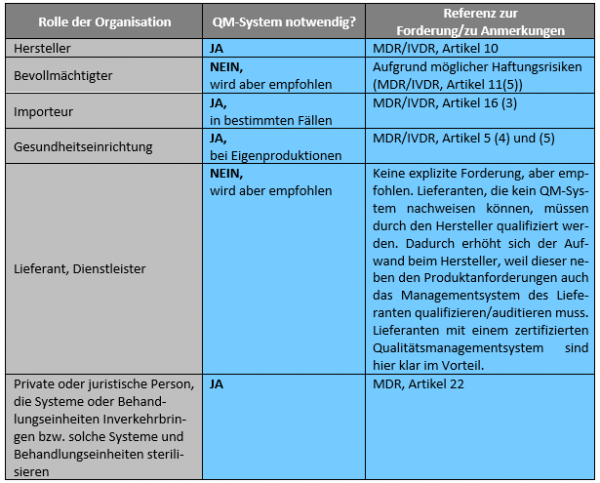
Wer benötigt ein QM System?
Neben den gesetzlich geregelten Rollen gem. MDR oder IVDR wird in der EN ISO 13485:2016 die Gruppe der Wirtschaftsakteure um weitere Rollen, wie z.B. Dienstleister, Zulieferer oder auch Spediteure er-gänzt. Nachdem diese Rollen außerhalb des gesetzlich geregelten Bereiches beschrieben werden, ist eine Implementierung eines Qualitätsmanagementsystems in solchen Organisationen freiwillig. Aus Gründen des Wettbewerbs oder spezieller Kundenanforderungen kann jedoch eine Implementierung eines Qualitätsmanagementsystems in diesen Unternehmen absolut sinnvoll sein.
Sie haben Fragen? Kontaktieren Sie uns! office@rmb-consulting.at
Fehler in der Technischen Dokumentation // Was Benannte Stellen dazu sagen

Wir haben uns bei Benannten Stellen schlau gemacht und ihnen entlocken können, welche Fehler der technischen Dokumentation (TD) häufig und für sie ein rotes Tuch sind.
Aktualität
Die TD ist nicht aktuell bzw. an MDR angepasst. Beispielsweise referenzieren Konformitätserklärung oder Gebrauchsanweisung auf die Richtlinie.
Lücken der Dokumentation
Häufig fehlen Querverweise innerhalb der TD, um eine Rückverfolgbarkeit ersichtlich darstellen zu können. Zusätzlich fehlt es oftmals an der Planung von Prüfungen inkl. der aussagekräftigen Bewertung der Ergebnisse. Die Frage, wie mit den Ergebnissen umgegangen wird, ist essentiell für die Qualität des Produktes!
GSLA - Grundlegende Sicherheits- & Leistungsanforderungen
Entweder diese werden nicht betrachtet oder der Nachweis enthält keine Referenzen auf eindeutige Nachweisdokumente. Hier ist auf Versionierung (Aktualität) der Nachweisdokumente zu achten!
Stand der Technik
Die Aktualität harmonisierter Normen VS. dem aktuellen Stand der Technik ist nicht ausreichend ermittelt.
Unzureichende Prozessbeschreibung
Prozesse - und deren Wechselwirkungen - sind unzureichend dargestellt. (Entwicklung, Produktion, Qualitätssicherungen, ausgelagerte Prozesse)
Keine Darstellung der Produkthistorie
Fehlende Dokumentation früherer Produktgenerationen.
Klinische Bewertung
Es wird nicht mit der aktuellen MEDDEV 2.7/1 rev. 4 gearbeitet. Bewertungen hinsichtlich PMCF fehlen.
PMS-Plan
Die Anforderungen aus MDR, Annex III sind nicht ausreichend umgesetzt.

IVDR - Geltungsbeginn
Durch die Umstellung von der IVDD zur IVDR werden aus 48 Seiten insgesamt 157 Seiten, dies bringt strengere regulatorische Vorschriften mit sich. Hersteller dürfen nicht vergessen, zeitgerecht eine Rezertifizierung ihrer Produkte vorzunehmen, auch wenn der Geltungsbeginn noch in der Ferne zu liegen scheint. Wie auch bei der MDR sollten die Hersteller die Umstellung nicht unterschätzen und als bald als möglich mit dem normkonformen Aufbau starten.
Ziel der neuen Verordnung ist die Stärkung von Schlüsselelemente des derzeitigen Regulierungskonzepts. Neu hinzu kommt die Einführung von Bestimmungen zur Gewährleistung von Transparenz und Rückverfolgbarkeit hinsichtlich einmaliger Produktkennung und der einmaligen Registrierungsnummer.
Am 25.05.17 ist die IVDR in Kraft getreten, es besteht eine Übergangsfrist von 5 Jahren bis zum 26.05.22. Ab diesem Zeitpunkt ist der Geltungsbeginn definiert. Bis dahin müssen auch noch Referenzlabore zur Verfügung gestellt werden (derzeit noch nicht erfolgt). Die benannten Stellen dürfen bis zum 26.05.22 noch Zertifikate nach IVDD ausstellen, diese sind dann längstens bis zum 27.05.24 gültig.
Die Erstinbetriebnahme von In vitro Diagnostik Produkte ist bis zum 27.05.25 gestattet. Danach müssen alle IVD Produkte nach der neuen IVD-Verordnung ausgerichtet sein. (Bestehende, bereits installierte und in Betrieb genommene IVDs dürfen weiterhin betrieben werden)

Produkte mit gültiger Zertifizierung unter IVDD dürfen nach dem 26. Mai 2022 nur in Verkehr gebracht oder erstmalig in Betrieb genommen werden, wenn folgende Punkte kumulativ gegeben sind:
- die IVDD Anforderungen müssen erfüllt sein
- es dürfen keine wesentlichen Änderungen am Produkt erfolgen
- eine Überwachung durch die benannte Stelle, die das Zertifikat unter IVDD ausgestellt hat, muss erfolgen
- folgende Anforderungen der IVDR müssen ab 26. Mai 2022 erfüllt werden:
- Registrierung Wirtschaftsakteure und Produkte
- Überwachung nach Inverkehrbringung (PMS)
- Vigilanz
Während der Übergangsfrist müssen alle CE gekennzeichneten Produkte nach IVD-Verordnung neubewertet werden.
Essenzielle Änderungen durch IVDR sind:
- ein neues Klassifizierungssystem (viele sog. Other Devices werden zukünftig in eine Klasse kommen, die eine Prüfung durch ein benannte Stelle notwendig macht!)
- Erweiterung des Geltungsbereichs und neue Wirtschaftsakteure
- Striktere Anforderungen an die Wirtschaftsakteure
- Strengere Überwachung - stärkere Mitwirkung der benannten Stellen
- Strengere Anforderungen an Transparenz und Rückverfolgbarkeit
- Strengere Richtlinien zur Technischen Dokumentation/klinischen Nachweis
- Strengere Richtlinien für Überwachung nach dem Inverkehrbringen
RnB-Consulting unterstützt Sie im Rahmen der Implementierung der europäischen Verordnungen für Medizinprodukte und In vitro Diagnostika (MDR und IVDR) und steht dabei mit erfahrenen Experten zur Seite.
Quelle: TÜV SÜD - modulare Weiterbildung - INHALTE UND UMSETZUNG DER NEUEN EUROPÄISCHEN IVD-VERORDNUNG
Persönliche Schutzausrüstung im Geschäfts- und Gesundheitsbereich
Anwendung, Einstufung, Zulassung und Kennzeichnung
Die Gesundheitsbedrohung durch COVID-19 stellt für Hersteller von Persönlicher Schutzausrüstung & Medizinprodukten sowie für die Anwender im Gesundheitsbereich aktuell eine Herausforderung dar.
Produkte zur persönlichen Schutzausrüstung (PSA) sind in der Europäischen Union (EU) grundlegend mit der sogenannten PSA-Verordnung (EU) Nr. 2016/425 geregelt. In Österreich gilt überdies die Verordnung Persönliche Schutzausrüstung1, welche eine Verordnung aus dem Arbeitnehmerschutzgesetz 2 darstellt.
Für Medizinprodukte (MP) im engeren Sinne gilt in der Europäischen Union die Medizinprodukte-Richtlinie 93/42/EWG, die national umgesetzt wurde. In Österreich sind die Vorgaben der Richtlinie im Medizinproduktegesetz3 verankert. Die Medizinprodukte-Richtline 93/42/EWG wird ab Mai 2021 durch die Medizinprodukteverordnung (EU) 2017/745 ersetzt. Diese ist bereits im Mai 2017 in Kraft getreten und hätte ihren Geltungsbeginn am 26. Mai 2020 gehabt. Aufgrund der Coronavirus-Krise wurde der Geltungsbeginn auf den 26. Mai 2021 verschoben.

Partikelfilternde Halbmasken sogenannte Filtering Face Piece - FFP-Masken - unterliegen der Verordnung für Persönliche Schutzausrüstung (EU) Nr. 2016/425. Sie bestehen vollständig aus Filtermaterial, sind meist vorgeformt oder aus mehreren Teilen zusammengesetzt. Sie umschließen den Mund und die Nase, sodass die Einatemluft durch den Filter strömen muss. Atemschutzmasken sind konzipiert, um den Träger vor festen und flüssigen Partikeln (Aerosole) zu schützen.
In Kontext zu COVID-19 bedeutet dies, dass der Träger sich gegen humane Tröpfchen- und Aerosol-Sekrete effektiv schützen kann, was somit eine wirksame Infektionsprävention gegen SARS-CoV-2 darstellt.
Der verbreitete Irrglaube, dass FFP-Masken im Allgemeinen auch das Gegenüber (Patienten) gegen die Tröpfchen- und Aerosol-Sekrete des Trägers schützen, ist nicht richtig. Nur wenn die FFP-Masken vom Hersteller speziell dafür ausgelegt wurden und auch als solches gekennzeichnet sind, ist ein Schutz von Träger und Patient gegeben. Diese FFP-Masken unterliegen dann der PSA-Verordnung und der nationalen Umsetzung der Medizinprodukte-Richtline und müssen als solche auch ausgewiesen werden. D.h. sie müssen beiden Rechtsnormen, PSA-Verordnung und nationale Umsetzung der Medizinprodukte-Richtline entsprechen.
Per Erlass des Bundesministeriums für Digitalisierung und Wirtschaftssandort dürfen in Österreich (und anderen EU-Ländern) auch sogenannte Corona SARS-Cov-2 Pandemie Atemschutzmasken (CPA) in Verkehr gebracht werden. Bei solchen CPA-Masken handelt es sich um FFP-Masken, welche in einem verkürzten Prüfverfahren ohne CE-Kennzeichnung in Österreich für medizinische Fachkräfte für die Dauer der derzeitigen COVID-Krise in Verkehr gebracht werden dürfen.

Medizinische Gesichtsmasken oder auch Medizinischer Mund-Nasen-Schutz genannt („OP-Maske“) unterliegen der nationalen Umsetzung der Medizinprodukterichtline und müssen als solche auch gekennzeichnet werden. Medizinische Gesichtsmasken bestehen meist aus einem zweilagigen Vliesstoff mit einem Filter in Form eines Tuches. Diese Gesichtsmasken wurden konzipiert, um Patienten präventiv vor Infektionen durch medizinisches Personal zu schützen.
Der verbreitete Irrglaube, dass dieser medizinische Mund-Nasen-Schutz den Träger wirksam gegen Tröpfchen- und Aerosol-Sekrete schützt, ist nicht richtig. Sie dienen dem Schutz des Patienten und nicht dem Schutz ihres Trägers.

Ist ein sogenannter „Mund-Nasen-Schutz“ (MNS) nicht als Medizinprodukt gekennzeichnet und ausgewiesen, handelt es sich nicht um einen medizinischen Mund-Nasen-Schutz mit einer definierten Schutzwirkung für den Patienten oder dem Gegenüber. Solch ein MNS wird im Allgemeinen als “Behelfs-Mund-Naseschutz” oder “Community-Maske” bezeichnet, welche keine definierte Schutzwirkung haben.

Gesichtsschutzschilder oder auch Face-Shield genannt unterliegen der Verordnung für persönliche Schutzausrüstung (EU) Nr. 2016/425. Die Schutzwirkung in der sozialen Konversation richtet sich primär gegen Tropfen und Spritzer von Flüssigkeiten.
In Kontext zu COVID-19 bedeutet dies, dass der Träger sich gegen humane Tröpfchen-Sekrete zu schützen versucht, was somit eine unterstützende Infektionsprävention gegen SARS-CoV-2 darstellt.
Wird vom Hersteller auch ein Patientenschutz angeführt („Medizinischer Gesichtsschutzschild“), welcher auch zum Schutz des Patienten dient, so handelt es sich auch um ein Medizinprodukt, welches unter die nationale Umsetzung der Medizinprodukte-Richtlinie (in Österreich das Medizinproduktegesetz) fällt. Dieser Gesichtsschutz muss somit dann beiden Rechtsnormen, PSA-Verordnung und der nationalen Umsetzung der Medizinprodukte-Richtlinie entsprechen.
Alle Medizinprodukte und persönliche Schutzausrüstungen müssen entsprechend CE-gekennzeichnet und die entsprechende Konformitätserklärung muss verfügbar sein. Die Marktaufsicht hinsichtlich der Medizinprodukte hat in Österreich das Bundesamt für Sicherheit im Gesundheitswesen (BASG) mit dem Geschäftsfeld Medizinmarktaufsicht der Agentur für Gesundheit und Ernährungssicherheit (AGES). Als Marktüberwachungsbehörde für persönliche Schutzausrüstung ist in Österreich die örtlich zuständige Bezirksverwaltungsbehörde zuständig.
Welche konkreten Präventionsmaßnahmen zum Mitarbeiter- und Kundenschutz gegen SARS-CoV-2 angebracht sind, muss von Fall zu Fall geprüft und evaluiert werden. Hierbei müssen die individuellen, spezifischen Betriebsabläufe entsprechend dem Infektionsrisiko bewertet und individuelle Präventionsmaßnahmen getroffen werden. Mit Erarbeitung eines entsprechenden Gesamtkonzeptes kann eine effiziente Infektionsprävention zum Schutz der Mitarbeiter und Kunden gegen SARS-CoV-2 erreicht werden.
Hierbei stehen wir Ihnen von Seiten RnB-Consulting mit unserem Expertenteam gerne zur Verfügung.
1Persönliche Schutzausrüstung (PSA-V) BGBl. II Nr. 77/2014
2ArbeitnehmerInnenschutzgesetz (ASchG) BGBl. I Nr. 450/1994
3Medizinproduktegesetz (MPG) BGBl. I Nr. 657/1996