Blog
Praxisbeispiel - FDA Approval für AI gestützte Diagnosesoftware
Am 11. April 2018 erhielt das diagnostische Medizinprodukt IDX-DR (IDx LLC, Iowa City,IA, USA) von der FDA die Freigabe für den US-Markt. Dies ist insbesonders interessant, da dieses Medizinprodukt als zentrales Element einen Algorithmus bereitstellt, der adaptiv gestaltet ist und auf Konzepten von künstlicher Intelligenz beruht. Diese Blog-Serie soll nun die unterschiedlichen Aspekte dieses Zulassungsverfahrens diskutieren – basierend auf öffentlich zugänglichen Informationen und Datenbanken. In diesem Zusammenhang werden wir beispielsweise Themen wie den De Novo Zulassungsprozess, Anforderungen an den Software-Lebenszyklus und Cybersecurity sowie die Strategie der klinischen Validierung und Bewertung diskutieren. Wir hoffen dadurch, Herstellern von ähnlichen Medizinprodukten ein Bild darüber geben zu können, wie die Zulassung dieser innovativen Algorithmen aus der regulatorischen Sicht der FDA durchgeführt werden kann. Wir werden uns für diese Recherche u.a. auf folgende Quellen beziehen:
- De Novo Classification Request for IDx-DR, 12. Jänner 2018
- Correspondence Letter der FDA, 11. April 2018
- Datenbankauszug von ClinicalTrials.gov (abgerufen am 27.12.2018)
- Klinisch-wissenschaftliche Publikationen zu dem Medizinprodukt
Zusätzlich werden wir an den entsprechenden Stellen auf relevante FDA Guidance Documents verweisen. Sämtliche Dokumente werden am Ende der jeweiligen Blog-Artikel referenziert.
Zweckbestimmung - Intended Use
Bevor wir uns im Detail mit dem Zulassungsverfahren und der klinischen Validierung beschäftigen, ist es natürlich notwendig, das vorliegende Medizinprodukt zu definieren und abzugrenzen. Das IDx-DR stellt eine Diagnosesoftware dar, welches für die automatisierte Detektion des Krankheitsbild diabetische Retinopathie eingesetzt wird. Dieses mit Diabetes assozierte Krankheitsbild kann durch eine zunehmende Schädigung der Blutgefäße in der Netzhaut bis zu Erblindung führen. Eine frühzeitige Erkennung dieser minimalen Schädigungen und ein entsprechende Einleitung therapeutischer Maßnahmen ist somit wichtig für den weiteren Krankheitsverlauf. Das IDx-DR analysiert nun Fundusaufnahmen der Netzhaut, um bei PatientInnen mit Diabetes, aber ohne bisheriger Diagnosestellung der diabetischen Retinopathie die nicht-milde Form dieses augenärztlichen Krankheitsbildes zu detektieren. Die therapeutische Konsequenz ist die Überweisung an Fachambulanzen und die mögliche Initiierung der Therapie (beispielsweise der intra-vitrealen Injektion von Arzneimitteln). Zusammenfassend kann das Software-System somit als Screening-Untersuchung angesehen werden (ein Aspekt, der bei der späteren Diskussion der klinischen Endpunkte und biometrischen Planung wieder aufgegriffen wird).
Beschreibung des Software-Systems
Die IDx-DR ist aktuell auf die gemeinsame Nutzung mit der Netzhautkamera Topcon NW400 eingeschränkt und kann in folgende Komponenten unterteilt werden:
- IDx-DR Client auf lokalen PC (Hochladen der Bilder, Empfang / Anzeige der Resultate…)
- IDx Web Service (Webserver Front-End zur Abarbeitung von Diagnoseanforderungen, Logging, Cybersecurity…)
- IDx-DR Analysis (analysiert Bilder hinsichtlich der Qualität und des Vorhandenseins von diabetischer Retinopathie)
Eine detailliertere Beschreibung der einzelnen Komponenten findet sich in dem De Novo Classification Request for IDx-DR auf den Seiten 2/3. Folgende Grafik wurde den Informationen in diesem Dokument nachgebildet.

Abgrenzung
- Die konkreten Therapieoptionen werden nicht durch die Diagnosesoftware vorgeschlagen und sollen an den entsprechenden Stellen ausschließlich dem klinischen Gesamtverständnis dienen.
- Die Netzhautkamera Topcon N400 selbst ist nicht Bestandteil des Medizinprodukts IDx-DR – wobei es natürlich Schnittstellen gibt.
Ausblick
Das Software-System wurde seitens der FDA die Kategorie Retinal diagnostic software device zugeordnet. Diese Kategorie und die entsprechende Regulation Number 21 CFR 886.1100 wurde als Ergebnis des De Novo Zulassungsprozesses neu geschaffen – näheres dazu im nächten Blog-Beitrag.
Referenzen
Wissen Sie wirklich was auf Sie zukommt?
RnB Consulting – „Es wird ernst!“

Sie sind Medizinprodukte-Hersteller und haben Ihr Produkt bereits erfolgreich auf den Markt gebracht? Dann schieben Sie die MDR/IVDR-Umstellung auf keinen Fall auf die lange Bank! Wenn Sie sich nicht bereits jetzt einen strukturierten „Fahrplan“ für die Umstellung Ihres Unternehmens und Ihrer Produkte machen, um den MDR-Anforderungen gerecht zu werden, könnte das im schlimmsten Fall das Aus für ihr Business bedeuten…
Medizinprodukte-Herstellern läuft die Zeit davon!
Die Übergangsfrist von drei Jahren hört sich im ersten Moment lang genug an, um interne Prozesse, Produktdokumentationen oder Zulassungsstrategien zu aktualisieren und an die Forderungen der EU-Verordnung anzupassen. Wenn Medizinprodukte-Hersteller jedoch nicht in absehbarer Zeit eine strukturierte Vorgehensweise der Umstellung definieren, könnte es für einige unter ihnen brenzlig werden.
RnB Consulting hat die Lösung!

Wir haben es uns zur persönlichen Aufgabe gemacht, Ihnen als Medizinprodukte-Hersteller bestmöglich mit Rat und Tat zur Seite zu stehen. Damit auch Sie den Anforderungen der EU-Verordnung – sei es MDR oder IVDR – gerecht werden, haben wir uns einen Masterplan überlegt, den wir speziell für Ihre Organisation und Ihre Produkte anbieten können.
 Phase 1: GAP-Analyse
Basierend auf Ihren aktuell zur Verfügung stehenden Dokumentationen - Anforderungen laut Richtlinie – erstellen wir eine GAP-Analyse inkl. Projektmanagement-Übersicht, um sich ein erstes Bild des Workloads „Umstellung MDD auf MDR“ machen zu können.
Phase 1: GAP-Analyse
Basierend auf Ihren aktuell zur Verfügung stehenden Dokumentationen - Anforderungen laut Richtlinie – erstellen wir eine GAP-Analyse inkl. Projektmanagement-Übersicht, um sich ein erstes Bild des Workloads „Umstellung MDD auf MDR“ machen zu können.
Phase 2: Angebotserstellung für ganzheitliche Unterstützung bei Umsetzung MDR/IVDR Sie wollen gemeinsam mit uns Durchstarten? Wir erstellen Ihnen gerne ein unverbindliches Angebot.
Phase 3: Los geht’s: Sie haben sich für uns als „helping hand“ entschieden? Wir nehmen die Herausforderung an! Anhand eines detaillierten Projektmanagements leiten wir Ihr „Umstellungsprojekt“, um gemeinsam ans Ziel zu kommen. Unsere Expertisen zu den unterschiedlichsten Fachdisziplinen, wie bspw. Qualitätsmanagement, klinischer Bewertung, Usability-Engineering, Software-Entwicklung & Dokumentation, werden Sie ganzheitlich betreuen und laufend für Sie da sein. Für weitere Fragen zu unseren Angeboten stehen wir Ihnen jederzeit sehr gerne zur Verfügung!
Der klinische Prüfbericht (Clinical Investigation Report – CIR)
Dieser Artikel beschreibt die Struktur, die Erstellung und die Kommunikation des klinischen Prüfberichts – einem essentiellen Nachweisdokument über die korrekte Abwicklung einer klinischen Prüfung.
Die Stärkung der klinischen Bewertung ist ein wesentliches Ziel der neuen EU-Verordnung für Medizinprodukte 1), welche nach Ablauf der Übergangsfrist spätestens am 26. Mai 2020 vollständig eingehalten werden muss. Der Nachweis des klinischen Leistungs- und Sicherheitsprofils muss hierbei typischerweise auf klinischen Daten basieren – hiervon sind nur sehr wenige Ausnahmen möglich. Die Tatsache, dass die Verwendung klinischer Daten von potentiellen Äquivalenzprodukten erschwert wird, hat bereits etliche Hersteller veranlasst, eigene klinische Prüfungen zu initiieren und Prozesse für die klinische Entwicklung ihrer Medizinprodukte zu definieren.
Ein Teil der Planung der klinischen Prüfung sollte hierbei auch die zu erstellenden Dokumente und deren Integration in die klinische Bewertung bzw. in die technische Dokumentation umfassen. Der klinische Prüfbericht bezeichnet hierbei ein zentrales Dokument, welches nach Beendigung der klinischen Prüfung erstellt werden muss. Dies betrifft sowohl die ordnungsgemäße als auch die vorgezogene Beendigung der klinischen Prüfung. Der Bericht stellt für Behördenvertreter, Ethikkommissionen, Gutachtern von benannten Stellen und zukünftig auch für die interessierte Öffentlichkeit eine wichtige Zusammenfassung der Merkmale und Ergebnisse der klinischen Prüfung dar.
An dieser Stelle ist anzumerken, dass der klinische Prüfbericht keinesfalls gleichzusetzen ist mit dem Bericht der klinischen Bewertung des Medizinprodukts (Clinical Evaluation Report – CER). Letzerer ist übergeordnet zu betrachten und referenziert als mögliche Quellen von klinischen Daten auf diese klinischen Prüfberichte aber auch auf Literaturrecherchen. Erwähnenswert ist zusätzlich, dass der klinische Prüfbericht zwar grundsätzlich, aber nicht im Detail, mit dem Aufbau einer typischen klinischen Publikation vergleichbar ist.

Bevor wir zu der geforderten Struktur des klinischen Prüfberichts übergehen, noch ein paar formale Aspekte. Der klinische Prüfbericht
- muss in schriftlicher Form vorliegen,
- darf ausschließlich anonymisierte klinische Daten enthalten, und
- muss sinnvollerweise von den beteiligten Personen unterschrieben sein (Sponsor, Prüfleiter, …).
Glücklicherweise orientiert sich die MDR 3) stark an der Strukturvorgabe der entsprechenden harmonisierten Norm EN ISO 14155 4), wobei auszugsweise folgende Punkte darzustellen sind:
- Deckblatt (Bezeichnung des Prüfproduktes, Bezeichnung der klinischen Prüfung / des klinischen Prüfplans, Name des Sponsors…)
- Zusammenfassung (kurze Beschreibung der klinischen Prüfung – Studiendesign, Ergebnisse, Schlussfolgerungen…)
- Einleitung (Zusammenhang zu klinischer Entwicklung, Studienziele, Fragestellungen…)
- Prüfprodukt und Prüfverfahren
- Beschreibung des Prüfprodukts, Änderungen am Prüfprodukt im Rahmen der Prüfungsdurchführung…
- Klinischer Prüfplan (Studiendesign, Ein-und Ausschlusskriterien, Studienhypothesen und Studienendpunkte, statistische Analyse, Monitoring, Verblindung…)
- Ergebnisse
- Deskriptive Darstellung der Charakteristika der ProbandInnen (Alter, Geschlecht…)
- Protokollabweichungen
- Analysen (Leistungsmerkmale, unerwünschte Ereignisse inklusive Produktmängel…)
- Vorzeitiger Abbruch von Versuchspersonen (Screening-Failures, Rücktritt…)
- Diskussionen und Gesamt-Schlussfolgerungen
- Diskussion bezüglich der Leistungsmerkmale (Wirksamkeit, Sicherheit)
- Gegenüberstellung von Nutzen und Risiken
- Abkürzungen und Definitionen
- Ethische Gesichtspunkte (Verweis auf Ethikvotum …)
- Prüfer und Verwaltungsstruktur der Prüfung
- Unterschriftenblatt
- Anhänge zum Bericht (Monitoring, Ethikkommissionen, Prüfleiter und Insitutionen…)
Die Referenzierung des klinischen Prüfberichts im Bericht der klinischen Bewertung erfolgt sinnvollerweise im Schritt 1 der klinischen Bewertung 5), der Identifikation der klinischen Daten. Wie klinische Daten aus anderen Quellen müssen die klinischen Daten der durchgeführten klinischen Prüfung(en) in weiterer Folge kritisch bewertet und analysiert, sowie im Kontext der entsprechenden grundlegenden Anforderungen betrachtet werden. Aus der Perspektive eines Gutachters muss selbstverständlich die Stringenz hinsichtlich der Ergebnisse und Schlussfolgerungen zwischen dem Bericht der klinischen Bewertung und des klinischen Prüfberichts gegeben sein. Zusätzlich erwartet werden typischerweise auch die komplementären Dokumente der klinischen Prüfung wie beispielsweise der klinische Prüfplan, Ethikvoten oder die Analyse der Übertragbarkeit bei internationalen Prüfungen. 6)
In Hinblick auf die Verordnung für Medizinprodukte und den klinischen Prüfbericht gilt es zusätzlich folgende Aspekte zu beachten 7):
- Mitteilungsfrist: typischerweise 1 Jahr bei ordnungsgemäßer Beendigung, 3 Monate bei vorzeitiger Beendigung (Ausnahmen mit Fristverlängerung über 1 Jahr mussten vorab im CIP wissenschaftlich begründet worden sein).
- Eine Zusammenfassung (Summary Report) in leicht verständlicher Sprache (orientiert sich am Anwender) muss verfasst werden (Leitlinien werden dazu noch von der Kommission definiert)
- Der klinische Prüfbericht und die Zusammenfassung werden in ein elektronisches System übertragen und öffentlich zugänglich gemacht.
Der nächste Beitrag wird sich mit den Anforderungen an den Bericht über die Leistungsstudie im Kontext der EU-Verordnung über In-vitro-Diagnostika (IVDR) beschäftigen.
Die R`n`B Consulting GmbH sowie die Mag. Andreas Raffeiner GmbH haben es sich zu Aufgabe gemacht, Hersteller von Medizinprodukten bei der Gestaltung, Durchführung, Aufzeichnung und Dokumentation von klinischen Prüfungen zu unterstützen. Gerne stehen wir als Auftragsprüfinstitut (CRO) zur Verfügung (www.rnb-consulting.at oder office@rnb-consulting.at).
Außerdem freut es uns ein innovatives Kooperationsprojekt ankündigen zu dürfen - das Competence Center for Medical Devices (mehr dazu unter www.ccmd.at).
Danke an Fr. Brigitte Raffeiner und Hrn. Dr. Wolfgang Ecker für die Unterstützung bei der Erstellung dieses Beitrags.
Mit R'n'B Consulting zur MDR-Compliance!
 Und nun ist es soweit - die Übergangsfrist von 3 Jahren (+1 Jahr aufgrund der Pandemie) sind vorüber!
Und nun ist es soweit - die Übergangsfrist von 3 Jahren (+1 Jahr aufgrund der Pandemie) sind vorüber!
Unsere Vorgehensweise
Egal ob Sie bereits Medizinprodukte am Markt haben oder eine künftige Inverkehrbringung anstreben - gemeinsam mit uns erarbeiten Sie einen „MDR-Fahrplan“ und erhalten eine Handlungsempfehlung mit welcher Sie mit Sicherheit ans Ziel kommen.
Von der IST-Stand-Analyse Ihrer aktuellen Compliance (MDD) bis hin zur MDR-Compliance
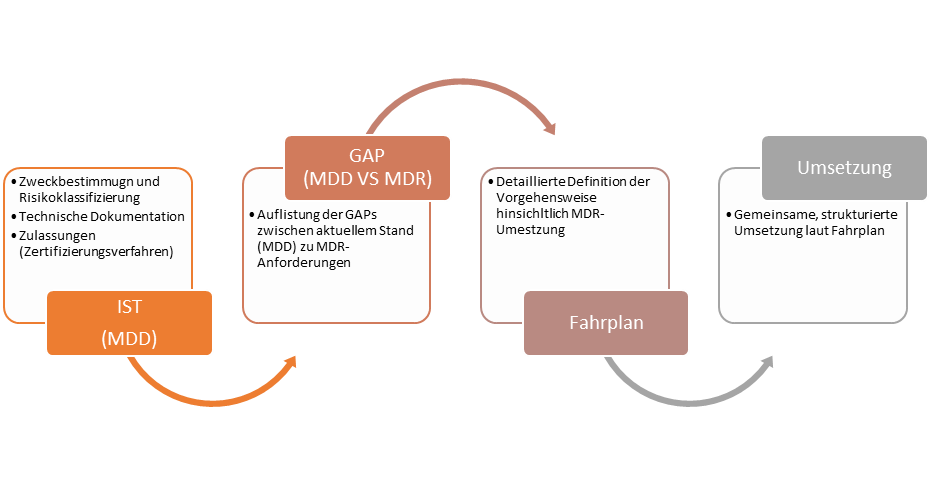
Wir haben Sie neugierig gemacht? Dann zögern Sie nicht länger und fragen Sie noch heute nach einem unverbindlichen, persönlichen Erstgespräch!
www.rnb-consulting.at
office@rnb-consulting.at
Erhebung klinischer Daten - Klinische Prüfung von Medizinprodukten
R'n'B Consulting und Mag. Andreas Raffeiner GmbH bieten einen ganzheitlichen Lösungsansatz
 Die Erhebung klinischer Daten ist für viele Hersteller von Medizinprodukten eine neuartige und große Herausforderung. Statistische Planungen und Analysen, das Verfassen von Studienprotokollen und die Durchführung von Einreichprozessen bei Ethikkommission und Behörden stellen hierbei nur einige Hürden dar.
Die Erhebung klinischer Daten ist für viele Hersteller von Medizinprodukten eine neuartige und große Herausforderung. Statistische Planungen und Analysen, das Verfassen von Studienprotokollen und die Durchführung von Einreichprozessen bei Ethikkommission und Behörden stellen hierbei nur einige Hürden dar.
Trigger für die Durchführung einer klinischen Prüfung kann beispielsweise ein systematisch identifizierter Mangel an klinischen Daten in Bezug auf spezielle Nebenwirkungen sein, welche nur schwierig für das eigene Produkt zu quantifizieren sind. So kann es beispielsweise bei speziellen orthopädischen Implantaten interessant sein, die Häufigkeit von Brüchen der Implantate über einen langen Zeitraum zu erheben. Bei periodisch stattfindender Auswertung besteht so zusätzlich auch die Möglichkeit einer Trendanalyse, welche eine wichtige und objektive Rückmeldung an das System der Marktüberwachung liefert.
Das erste Arbeitspaket in Hinblick auf die Durchführung klinischer Prüfung umfasst typischerweise die Definition der Eckdaten: die zu erhebenden medizinischen Parameter, die zugehörigen Zeitpunkte der Erhebung, das Studiendesign und die Anzahl der Studienzentren und geplanten ProbandInnen müssen wohl überlegt und begründbar sein. Normative Grundlage für die Durchführung von klinischen Prüfungen an Menschen ist der harmonisierte Standard EN ISO 14155. Neben etlichen wichtigen Definitionen ist darin beispielsweise auch die erwartete Struktur des klinischen Prüfplans festgelegt, welches das Rahmenwerk für die klinische Prüfung bildet.
Gemeinsam mit Begleitdokumenten muss dieser klinsische Prüfplan in weiterer Folge extern begutachtet werden. In Abhängigkeit der Medizinprodukte-Klassifizierung, des Zulassungsstatus und der Art der Anwendung (langfristig, invasiv) gibt es hierbei beachtliche Unterschiede hinsichtlich der Einreichmodalitäten unter Miteinbeziehung der Ethikkommission und Behörde. Die Studiendurchführung selbst stellt spezielle Herausforderungen an das Prüfteam vor Ort. Bei der Initiierungsvisite bereitet der Projektmanager und/oder Monitor das Projekt mit Hilfs- und Unterstützungsmaterialien so auf, dass auch unter Alltagsbedingungen eine protokollkonforme Abwicklung und fehlerfreie Dokumentation möglich ist.
Regelmäßiges unabhängiges Monitoring im Sinne von Begleiten und Unterstützen, sowie Überwachen der gesetzlichen Anforderungen ist ein unverzichtbares Element der Qualitätssicherung und –kontrolle im Laufe der konkreten Studiendurchführung.
Die erhobenen klinischen Daten werden vom Prüfteam in elektronische Datenaufzeichnungsbögen (eCRFs) eingetragen, die spezielle Anforderungen erfüllen müssen. Dabei ist es wichtig, dass diese Systeme validiert, aber auch einfach und intuitiv zu bedienen sind.
 Monitore helfen auch bei den gesetzlich vorgegebenen Meldeverpflichtungen von Unerwünschten Ereignissen (AEs) und Schwerwiegenden Unerwünschten Ereignissen (SAEs), die innerhalb eines Zeitfensters (24 Stunden im Fall von SAEs) gemeldet werden müssen.
Zeitersparnis und Komfort ist dann gegeben, wenn diese Meldeverpflichtungen auch in die eCRFs direkt eingetragen werden können.
Monitore helfen auch bei den gesetzlich vorgegebenen Meldeverpflichtungen von Unerwünschten Ereignissen (AEs) und Schwerwiegenden Unerwünschten Ereignissen (SAEs), die innerhalb eines Zeitfensters (24 Stunden im Fall von SAEs) gemeldet werden müssen.
Zeitersparnis und Komfort ist dann gegeben, wenn diese Meldeverpflichtungen auch in die eCRFs direkt eingetragen werden können.
Nach dem Abschluss der klinischen Prüfung und der statistischen Auswertung fließen die erhobenen klinischen Daten nun typischerweise über das Risikomanagement in den Bericht der klinischen Bewertung ein und stellen eine relevante und belastbare Datenquelle dar.
Medizinprodukte und der geforderte hohe Qualitätsstandard gehen als untrennbare Einheit Hand in Hand. Die Partnerunternehmen RnB Consulting und Mag. Andreas Raffeiner GmbH sind erfahrene Spezialisten bei der Projektabwicklung und im Datenmanagement.
Maßgeschneiderte und kompetente Lösungen sind dabei ein Gebot der Stunde. Wir unterstützen Sie bei Ihren Projekten.
- @ office@rnb-consulting.at
- ☎ +43 (0) 677 6122 89 67
- @ office@studien-monitor.at
- ☎ +43 7234 83764
<< Newer entries | Older entries >>